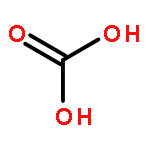
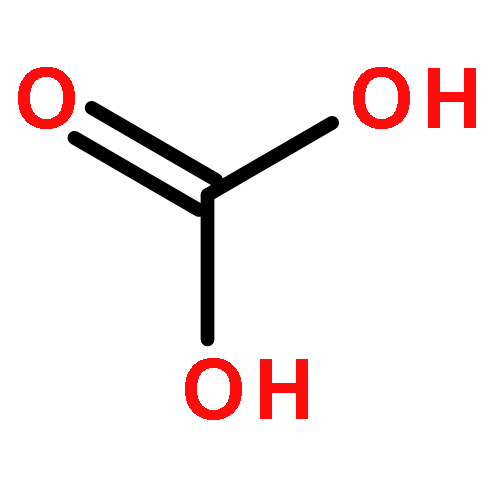
Co-reporter:Jacob W. Smith and Richard J. Saykally
Chemical Reviews December 13, 2017 Volume 117(Issue 23) pp:13909-13909
Publication Date(Web):November 10, 2017
DOI:10.1021/acs.chemrev.7b00213
X-ray absorption spectroscopy (XAS) is an electronic absorption technique for which the initial state is a deeply buried core level. The photon energies corresponding to such transitions are governed primarily by the binding energies of the initial state. Because the binding energies of core electrons vary significantly among atomic species, this makes XAS an element-selective spectroscopy. Proper interpretation of XA spectra can provide detailed information on the local chemical and geometric environment of the target atom. The introduction of liquid microjet and flow cell technologies into XAS experiments has enabled the general study of liquid samples. Liquids studied to date include water, alcohols, and solutions with relevance to biology and energy technology. This Review summarizes the experimental techniques employed in XAS studies of liquid samples and computational methods used for interpretation of the resulting spectra and summarizes salient experiments and results obtained in the XAS investigations of liquids.
Co-reporter:Debra L. McCaffrey;Son C. Nguyen;Stephen J. Cox;Horst Weller;A. Paul Alivisatos;Phillip L. Geissler
PNAS 2017 114 (51 ) pp:13369-13373
Publication Date(Web):2017-12-19
DOI:10.1073/pnas.1702760114
The adsorption of ions to aqueous interfaces is a phenomenon that profoundly influences vital processes in many areas of science,
including biology, atmospheric chemistry, electrical energy storage, and water process engineering. Although classical electrostatics
theory predicts that ions are repelled from water/hydrophobe (e.g., air/water) interfaces, both computer simulations and experiments
have shown that chaotropic ions actually exhibit enhanced concentrations at the air/water interface. Although mechanistic
pictures have been developed to explain this counterintuitive observation, their general applicability, particularly in the
presence of material substrates, remains unclear. Here we investigate ion adsorption to the model interface formed by water
and graphene. Deep UV second harmonic generation measurements of the SCN− ion, a prototypical chaotrope, determined a free energy of adsorption within error of that for air/water. Unlike for the
air/water interface, wherein repartitioning of the solvent energy drives ion adsorption, our computer simulations reveal that
direct ion/graphene interactions dominate the favorable enthalpy change. Moreover, the graphene sheets dampen capillary waves
such that rotational anisotropy of the solute, if present, is the dominant entropy contribution, in contrast to the air/water
interface.
Co-reporter:Debra L. McCaffrey;Son C. Nguyen;Stephen J. Cox;Horst Weller;A. Paul Alivisatos;Phillip L. Geissler
PNAS 2017 114 (51 ) pp:13369-13373
Publication Date(Web):2017-12-19
DOI:10.1073/pnas.1702760114
The adsorption of ions to aqueous interfaces is a phenomenon that profoundly influences vital processes in many areas of science,
including biology, atmospheric chemistry, electrical energy storage, and water process engineering. Although classical electrostatics
theory predicts that ions are repelled from water/hydrophobe (e.g., air/water) interfaces, both computer simulations and experiments
have shown that chaotropic ions actually exhibit enhanced concentrations at the air/water interface. Although mechanistic
pictures have been developed to explain this counterintuitive observation, their general applicability, particularly in the
presence of material substrates, remains unclear. Here we investigate ion adsorption to the model interface formed by water
and graphene. Deep UV second harmonic generation measurements of the SCN− ion, a prototypical chaotrope, determined a free energy of adsorption within error of that for air/water. Unlike for the
air/water interface, wherein repartitioning of the solvent energy drives ion adsorption, our computer simulations reveal that
direct ion/graphene interactions dominate the favorable enthalpy change. Moreover, the graphene sheets dampen capillary waves
such that rotational anisotropy of the solute, if present, is the dominant entropy contribution, in contrast to the air/water
interface.
Co-reporter:Debra L. McCaffrey;Son C. Nguyen;Stephen J. Cox;Horst Weller;A. Paul Alivisatos;Phillip L. Geissler
PNAS 2017 114 (51 ) pp:13369-13373
Publication Date(Web):2017-12-19
DOI:10.1073/pnas.1702760114
The adsorption of ions to aqueous interfaces is a phenomenon that profoundly influences vital processes in many areas of science,
including biology, atmospheric chemistry, electrical energy storage, and water process engineering. Although classical electrostatics
theory predicts that ions are repelled from water/hydrophobe (e.g., air/water) interfaces, both computer simulations and experiments
have shown that chaotropic ions actually exhibit enhanced concentrations at the air/water interface. Although mechanistic
pictures have been developed to explain this counterintuitive observation, their general applicability, particularly in the
presence of material substrates, remains unclear. Here we investigate ion adsorption to the model interface formed by water
and graphene. Deep UV second harmonic generation measurements of the SCN− ion, a prototypical chaotrope, determined a free energy of adsorption within error of that for air/water. Unlike for the
air/water interface, wherein repartitioning of the solvent energy drives ion adsorption, our computer simulations reveal that
direct ion/graphene interactions dominate the favorable enthalpy change. Moreover, the graphene sheets dampen capillary waves
such that rotational anisotropy of the solute, if present, is the dominant entropy contribution, in contrast to the air/water
interface.
Co-reporter:Debra L. McCaffrey;Son C. Nguyen;Stephen J. Cox;Horst Weller;A. Paul Alivisatos;Phillip L. Geissler
PNAS 2017 114 (51 ) pp:13369-13373
Publication Date(Web):2017-12-19
DOI:10.1073/pnas.1702760114
The adsorption of ions to aqueous interfaces is a phenomenon that profoundly influences vital processes in many areas of science,
including biology, atmospheric chemistry, electrical energy storage, and water process engineering. Although classical electrostatics
theory predicts that ions are repelled from water/hydrophobe (e.g., air/water) interfaces, both computer simulations and experiments
have shown that chaotropic ions actually exhibit enhanced concentrations at the air/water interface. Although mechanistic
pictures have been developed to explain this counterintuitive observation, their general applicability, particularly in the
presence of material substrates, remains unclear. Here we investigate ion adsorption to the model interface formed by water
and graphene. Deep UV second harmonic generation measurements of the SCN− ion, a prototypical chaotrope, determined a free energy of adsorption within error of that for air/water. Unlike for the
air/water interface, wherein repartitioning of the solvent energy drives ion adsorption, our computer simulations reveal that
direct ion/graphene interactions dominate the favorable enthalpy change. Moreover, the graphene sheets dampen capillary waves
such that rotational anisotropy of the solute, if present, is the dominant entropy contribution, in contrast to the air/water
interface.
Co-reporter:Anthony M. RizzutoErik S. Cheng, Royce K. Lam, Richard J. Saykally
The Journal of Physical Chemistry C 2017 Volume 121(Issue 8) pp:
Publication Date(Web):February 6, 2017
DOI:10.1021/acs.jpcc.6b12851
The kinetics and energetics of cloud droplet and aerosol formation in the atmosphere are strongly influenced by the evaporation and condensation rates of water, yet the magnitude and mechanism of evaporation remains incompletely characterized. Of particular import (and controversy) is the nature of interfacial water pH and its potential effects on evaporation rate and environmental reactivity. We have used Raman thermometry measurements of freely evaporating microdroplets to determine evaporation coefficients (γ) for two different hydrochloric acid solutions, both of which result in a significant deviation from γwater. At 95% confidence, we find the evaporation coefficient for 1.0 M HCl to be 0.24 ± 0.04, a ∼60% decrease relative to pure water, and for 0.1 M HCl to be 0.91 ± 0.08, a ∼45% increase relative to pure water. These results suggest a large perturbation in the surface structure induced by either hydronium ions adsorbing to the water surface or by the presence of a Cl–···H3O+ ion-pair moiety in the interfacial region.
Co-reporter:William T. S. Cole;James D. Farrell;David J. Wales
Science 2016 Vol 352(6290) pp:1194-1197
Publication Date(Web):03 Jun 2016
DOI:10.1126/science.aad8625
A close-up look at eight water molecules
A raindrop may look small, but it contains far too much water to model with the highest chemical precision. Theorists rely on studies of clusters with just a few molecules to enhance their understanding of the quantum-mechanical forces at play in the liquid. Cole et al. now report a high-resolution spectrum in the terahertz regime of the eight-membered cluster. By resolving 99 absorption lines associated with a collective torsional mode, the authors distinguish prolate and oblate isomers that are very similar in energy.
Science, this issue p. 1194
Co-reporter:Nadine Schwierz
The Journal of Physical Chemistry C 2016 Volume 120(Issue 27) pp:14513-14521
Publication Date(Web):June 9, 2016
DOI:10.1021/acs.jpcc.6b03788
Liquid water microjets have been successfully employed for both electrical power generation and gaseous hydrogen production, but the demonstrated efficiencies have been low. Here, we employ a combination of a modified Poisson–Boltzmann description, continuum hydrodynamic equations, and microjet electrokinetic experiments to gain detailed insight into the origin of the streaming currents produced in pure water. We identify the contributions to the streaming current from specific ion adsorption at the solid/liquid interface and from long-ranged electrostatic interactions, finding that the portion originating from the latter dominate at charged surfaces. The detailed understanding afforded by theory and the close agreement with experimental results elucidates design principles for optimizing hydrogen production and power generation. Changing the sign of the surface charge density through targeted use of surface coatings via silanization switches the primary charge carrier between hydronium and hydroxide and therefore switches the corresponding production of molecular hydrogen to oxygen at the target electrode. Moreover, hydrophobic surface coatings reduce dissipation due to fluid/solid friction, thereby increasing the conversion efficiency.
Co-reporter:Royce K. Lam, Alice H. England, Jacob W. Smith, Anthony M. Rizzuto, Orion Shih, David Prendergast, Richard J. Saykally
Chemical Physics Letters 2015 Volume 633() pp:214-217
Publication Date(Web):16 July 2015
DOI:10.1016/j.cplett.2015.05.039
Highlights
- •
First X-ray absorption spectrum of dissolved CO2.
- •
Fast-flow liquid microjet mixing system used to generate dissolved CO2 via the decomposition of aqueous carbonic acid.
- •
Hydration structure determined in conjunction with first principles XCH calculations.
Co-reporter:William T.S. Cole, Nik C. Hlavacek, Alan W.M. Lee, Tsung-Yu Kao, Qing Hu, John L. Reno, Richard J. Saykally
Chemical Physics Letters 2015 Volume 638() pp:144-148
Publication Date(Web):1 October 2015
DOI:10.1016/j.cplett.2015.08.027
Highlights
- •
Design of THz VRT spectrometer utilizing quantum cascade lasers.
- •
Application of spectrometer to pulsed supersonic expansions.
- •
Comparison of QCL system to tunable FIR sideband spectrometer.
- •
∼1 ppm absorption sensitivity and ∼1 ppm frequency resolution.
Co-reporter:Jacob W. Smith, Royce K. Lam, Alex T. Sheardy, Orion Shih, Anthony M. Rizzuto, Oleg Borodin, Stephen J. Harris, David Prendergast and Richard J. Saykally
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 43) pp:23568-23575
Publication Date(Web):20 Aug 2014
DOI:10.1039/C4CP03240C
Since their introduction into the commercial marketplace in 1991, lithium ion batteries have become increasingly ubiquitous in portable technology. Nevertheless, improvements to existing battery technology are necessary to expand their utility for larger-scale applications, such as electric vehicles. Advances may be realized from improvements to the liquid electrolyte; however, current understanding of the liquid structure and properties remains incomplete. X-ray absorption spectroscopy of solutions of LiBF4 in propylene carbonate (PC), interpreted using first-principles electronic structure calculations within the eXcited electron and Core Hole (XCH) approximation, yields new insight into the solvation structure of the Li+ ion in this model electrolyte. By generating linear combinations of the computed spectra of Li+-associating and free PC molecules and comparing to the experimental spectrum, we find a Li+–solvent interaction number of 4.5. This result suggests that computational models of lithium ion battery electrolytes should move beyond tetrahedral coordination structures.
Co-reporter:Royce K. Lam, Alice H. England, Alex T. Sheardy, Orion Shih, Jacob W. Smith, Anthony M. Rizzuto, David Prendergast, Richard J. Saykally
Chemical Physics Letters 2014 Volume 614() pp:282-286
Publication Date(Web):20 October 2014
DOI:10.1016/j.cplett.2014.09.052
Highlights
- •
First X-ray absorption spectrum of aqueous carbonic acid.
- •
Fast-flow liquid jet mixing system used to generate the short lived acid.gauche- to syn-chloroacetone.
- •
Acid hydration structure determined in conjunction with first principles XCH theory.
Co-reporter:Wei Lin, David W. Steyert, Nikolaus C. Hlavacek, Anamika Mukhopadhyay, Ralph H. Page, Peter H. Siegel, Richard J. Saykally
Chemical Physics Letters 2014 Volume 612() pp:167-171
Publication Date(Web):18 September 2014
DOI:10.1016/j.cplett.2014.08.019
Highlights
- •
Over 600 VRT transitions measured.
- •
Proposed structure of dimer presented.
- •
Over 250 transitions fit to detailed Hamiltonian.
Co-reporter:Kaitlin C. Duffey, Orion Shih, Nolan L. Wong, Walter S. Drisdell, Richard J. Saykally and Ronald C. Cohen
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 28) pp:11634-11639
Publication Date(Web):07 May 2013
DOI:10.1039/C3CP51148K
The presence of organic surfactants in atmospheric aerosol may lead to a depression of cloud droplet growth and evaporation rates affecting the radiative properties and lifetime of clouds. Both the magnitude and mechanism of this effect, however, remain poorly constrained. We have used Raman thermometry measurements of freely evaporating micro-droplets to determine evaporation coefficients for several concentrations of acetic acid, which is ubiquitous in atmospheric aerosol and has been shown to adsorb strongly to the air–water interface. We find no suppression of the evaporation kinetics over the concentration range studied (1–5 M). The evaporation coefficient determined for 2 M acetic acid is 0.53 ± 0.12, indistinguishable from that of pure water (0.62 ± 0.09).
Co-reporter:Daniel N. Kelly, Royce K. Lam, Andrew M. Duffin, and Richard J. Saykally
The Journal of Physical Chemistry C 2013 Volume 117(Issue 24) pp:12702-12706
Publication Date(Web):May 30, 2013
DOI:10.1021/jp403583r
We describe a novel method that exploits electrokinetic streaming current measurements for the study of ion-interface affinity. Through the use of liquid microjets and ultradilute solutions (<1 μM), we are able to overcome inherent difficulties in electrokinetic surface measurements engendered by changing double-layer thicknesses. Varying bulk KCl concentrations produce statistically significant changes in streaming current down at picomolar concentrations. Because the attending ion concentrations are below that from water autoionization, these data are compared with those from ultradilute HCl and KOH solutions assuming that the K+ and Cl– introduce no new counterions. This permits comparison of the individual effects of K+ and Cl– on the interface, evidencing a cooperative effect between these ions at silica surfaces. Altogether, these results establish the effectiveness of this experimental approach in revealing new ion–surface phenomena and indicate its promise for the general study of aqueous interfaces.
Co-reporter:Jeremy O. Richardson, David J. Wales, Stuart C. Althorpe, Ryan P. McLaughlin, Mark R. Viant, Orion Shih, and Richard J. Saykally
The Journal of Physical Chemistry A 2013 Volume 117(Issue 32) pp:6960-6966
Publication Date(Web):January 3, 2013
DOI:10.1021/jp311306a
We report a combined theoretical and experimental study of the water octamer-h16. The calculations used the ring-polymer instanton method to compute tunnelling paths and splittings in full dimensionality. The experiments measured extensive high resolution spectra near 1.4 THz, for which isotope dilution experiments and group theoretical analysis support assignment to the octamer. Transitions appear as singlets, consistent with the instanton paths, which involve the breakage of two hydrogen-bonds and thus give tunneling splittings below experimental resolution.
Co-reporter:D.E. Otten, R. Onorato, R. Michaels, J. Goodknight, R.J. Saykally
Chemical Physics Letters 2012 s 519–520() pp: 45-48
Publication Date(Web):
DOI:10.1016/j.cplett.2011.10.056
Co-reporter:Andrew M. Duffin, Alice H. England, Craig P. Schwartz, Janel S. Uejio, Gregory C. Dallinger, Orion Shih, David Prendergast and Richard J. Saykally
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 38) pp:17077-17083
Publication Date(Web):2011/08/05
DOI:10.1039/C1CP21788G
Borohydride salts have been considered as good prospects for transportable hydrogen storage materials, with molecular hydrogen released via hydrolysis. We examine details of the hydration of sodium borohydride by the combination of X-ray absorption spectroscopy and first principles' theory. Compared to solid sodium borohydride, the aqueous sample exhibits an uncharacteristically narrow absorption feature that is shifted to lower energy, and ascribed to the formation of dihydrogen bonds between borohydride and water that weaken the boron–hydrogen covalent bonds. Water also acts to localize the highly excited molecular orbitals of borohydride, causing transitions to excited states with p character to become more intense and a sharp feature, uncharacteristic of tetrahedral molecules, to emerge. The simulations indicate that water preferentially associates with borohydride on the tetrahedral corners and edges.
Co-reporter:Alice H. England, Andrew M. Duffin, Craig P. Schwartz, Janel S. Uejio, David Prendergast, Richard J. Saykally
Chemical Physics Letters 2011 Volume 514(4–6) pp:187-195
Publication Date(Web):6 October 2011
DOI:10.1016/j.cplett.2011.08.063
Abstract
The dissolution of carbon dioxide in water and the ensuing hydrolysis reactions are of profound importance for understanding the behavior and control of carbon in the terrestrial environment. The first X-ray absorption spectra of aqueous carbonate have been measured at three different pH values to characterize the evolution of electronic structure of carbonate, bicarbonate, carbonic acid and dissolved CO2. The corresponding carbon K-edge core-level spectra were calculated using a first-principles electronic structure approach which samples molecular dynamics trajectories. Measured and calculated spectra are in excellent agreement. Each species exhibits similar, but distinct, spectral features which are interpreted in terms of the relative C–O bond strengths, molecular configuration, and hydration strength.
Co-reporter:Abigail E. Miller, Poul B. Petersen, Christopher W. Hollars, and Richard J. Saykally, Jan Heyda and Pavel Jungwirth
The Journal of Physical Chemistry A 2011 Volume 115(Issue 23) pp:5873-5880
Publication Date(Web):March 17, 2011
DOI:10.1021/jp110103j
The adsorption and aggregation of β-amyloid (1−16) fragment at the air−water interface was investigated by the combination of second harmonic generation (SHG) spectroscopy, Brewster angle microscopy (BAM), and molecular dynamics simulations (MD). The Gibbs free energy of surface adsorption was measured to be −10.3 kcal/mol for bulk pHs of 7.4 and 3, but no adsorption was observed for pH 10−11. The 1−16 fragment is believed not to be involved in fibril formation of the β-amyloid protein, but it exhibits interesting behavior at the air−water interface, as manifested in two time scales for the observed SHG response. The shorter time scale (minutes) reflects the surface adsorption, and the longer time scale (hours) reflects rearrangement and aggregation of the peptide at the air−water interface. Both of these processes are also evidenced by BAM measurements. MD simulations confirm the pH dependence of surface behavior of the β-amyloid, with largest surface affinity found at pH = 7. It also follows from the simulations that phenylalanine is the most surface exposed residue, followed by tyrosine and histidine in their neutral form.
Co-reporter:Craig P. Schwartz, Janel S. Uejio, Andrew M. Duffin, Walter S. Drisdell, Jared D. Smith, Richard J. Saykally
Chemical Physics Letters 2010 Volume 493(1–3) pp:94-96
Publication Date(Web):17 June 2010
DOI:10.1016/j.cplett.2010.05.037
X-ray absorption spectra of 1 M aqueous solutions of indium(III) chloride, yttrium(III) bromide, lanthanum(III) chloride, tin(IV) chloride and chromium(III) chloride have been measured at the oxygen K-edge. Relatively minor changes are observed in the spectra compared to that of pure water. SnCl4 and CrCl3 exhibit a new onset feature, which in the case of SnCl4 can probably be attributed to formation of hydroxide or other complex molecules in the solution. At higher energy, only relatively minor, but salt-specific changes in the spectra occur. The small magnitude of the observed spectral changes is ascribed to offsetting perturbations by the cations and anions.The oxygen K-edge NEXAFS spectrum of bulk water (black, solid) and 1 M solutions of SnCl4 (purple,-·-·) and CrCl3 (yellow,···) in the pre-edge region shows large spectral changes with salt identity.
Co-reporter:Robert M. Onorato, Dale E. Otten and Richard J. Saykally
The Journal of Physical Chemistry C 2010 Volume 114(Issue 32) pp:13746-13751
Publication Date(Web):July 2, 2010
DOI:10.1021/jp103454r
Recent experimental and theoretical work has demonstrated that certain anions can exhibit enhanced concentrations at aqueous interfaces and that the adsorption of bromide is particularly important for chemical reactions on atmospheric aerosols, including the depletion of ozone. UV second harmonic generation resonant with the bromide charge-transfer-to-solvent band and a Langmuir adsorption model are used to determine the affinity of bromide for both the air/water and dodecanol/water interfaces. The Gibbs free energy of adsorption for the former is determined to be −1.4 kJ/mol with a lower 90% confidence limit of −4.1 kJ/mol. For the dodecanol/water interface the data are best fit with a Gibbs free energy of +8 kJ/mol with an estimated lower limit of −4 kJ/mol.
Co-reporter:Janel S. Uejio;Alice H. England;Craig P. Schwartz;Andrew M. Duffin;David Prendergast;Daniel N. Kelly
PNAS 2010 Volume 107 (Issue 32 ) pp:14008-14013
Publication Date(Web):2010-08-10
DOI:10.1073/pnas.1006435107
Nitrogen K-edge spectra of aqueous triglycine were measured using liquid microjets, and the effects of Hofmeister-active salts
on the spectra were observed. Spectra simulated using density functional theory, sampled from room temperature classical molecular
dynamics trajectories, capture all major features in the measured spectra. The spectrum of triglycine in water is quite similar
to that in the presence of chaotropic sodium bromide (and other halides), which raises the solubility of proteins. However,
a new feature is found when kosmotropic Na2SO3, which lowers solubility, is present; this feature results from excitations of the nitrogen atom in the terminal amino group
of triglycine. Both direct interactions between this salt and the protonated amino terminus, as well as corresponding changes
in the conformational dynamics of the system, contribute to this new feature. These molecular measurements support a different
mechanism for the Hofmeister effect than has previously been suggested based on thermodynamic measurements. It is also shown
that near edge X-ray absorption fine structure (NEXAFS) is sensitive to strong direct interaction between certain salts and
charged peptides. However, by investigating the sensitivity of NEXAFS to the extreme structural differences between model
β-sheets and α-helices, we conclude that this technique is relatively insensitive to secondary structure of peptides and proteins.
Co-reporter:Walter S. Drisdell, Richard J. Saykally and Ronald C. Cohen
The Journal of Physical Chemistry C 2010 Volume 114(Issue 27) pp:11880-11885
Publication Date(Web):May 25, 2010
DOI:10.1021/jp101726x
Current understanding of the vapor−liquid exchange kinetics of liquid water is incomplete, leading to uncertainties in modeling the climatic effects of clouds and aerosol. Initial studies of atmospherically relevant solutes (ammonium sulfate, sodium chloride) indicate that their effect on the evaporation kinetics of water is minimal, but all those constituent ions are also expected to be depleted in concentration at the air−water interface. We present measurements of the evaporation kinetics of water from 4 M sodium perchlorate solution, which is expected to have an enhanced concentration of perchlorate in the surface layer, using Raman thermometry of liquid microdroplets in a free evaporation regime. We determine the evaporation coefficient γe to be 0.47 ± 0.02, ca. 25% smaller than our measured value for pure water (0.62 ± 0.09). This change, while small, indicates that direct interactions between perchlorate ions and evaporating water molecules are affecting the evaporation mechanism and kinetics and suggests that other solutes with high surface affinities may also produce a similar influence in the atmosphere and elsewhere.
Co-reporter:Paul Peng, Bryce Sadtler, A. Paul Alivisatos and Richard J. Saykally
The Journal of Physical Chemistry C 2010 Volume 114(Issue 13) pp:5879-5885
Publication Date(Web):March 4, 2010
DOI:10.1021/jp9116722
Electron relaxation dynamics in CdS−Ag2S nanorods have been measured as a function of the relative fraction of the two semiconductors, which can be tuned via cation exchange between Cd2+ and Ag+. The transient bleach of the first excitonic state of the CdS nanorods is characterized by a biexponential decay corresponding to fast relaxation of the excited electrons into trap states. This signal completely disappears when the nanorods are converted to Ag2S but is fully recovered after a second exchange to convert them back to CdS, demonstrating annealing of the nonradiative trap centers probed and the robustness of the cation exchange reaction. Partial cation exchange produces heterostructures with embedded regions of Ag2S within the CdS nanorods. Transient bleaching of the CdS first excitonic state shows that increasing the fraction of Ag2S produces a greater contribution from the fast component of the biexponential bleach recovery, indicating that new midgap relaxation pathways are created by the Ag2S material. Transient absorption with a mid-infrared probe further confirms the presence of states that preferentially trap electrons on a time scale of 1 ps, 2 orders of magnitude faster than that of the parent CdS nanorods. These results suggest that the Ag2S regions within the heterostucture provide an efficient relaxation pathway for excited electrons in the CdS conduction band.
Co-reporter:Andrew M. Duffin and Richard J. Saykally
The Journal of Physical Chemistry C 2008 Volume 112(Issue 43) pp:17018-17022
Publication Date(Web):October 8, 2008
DOI:10.1021/jp8015276
Although electrokinetic effects are not new, only recently have they been investigated for possible use in energy conversion devices. We recently reported the electrokinetic generation of molecular hydrogen from rapidly flowing liquid water microjets [Duffin et al. J. Phys. Chem. C 2007, 111, 12031]. Here, we describe the use of liquid water microjets for direct conversion of electrokinetic energy to electrical power. Previous studies of electrokinetic power production have reported low efficiencies (∼3%), limited by back conduction of ions at the surface and in the bulk liquid. Liquid microjets eliminate energy dissipation due to back conduction and, measuring only at the jet target, yield conversion efficiencies exceeding 10%.
Co-reporter:Janel S. Uejio;Craig P. Schwartz;Andrew M. Duffin;Walter S. Drisdell;Ronald C. Cohen
PNAS 2008 105 (19 ) pp:6809-6812
Publication Date(Web):2008-05-13
DOI:10.1073/pnas.0800181105
We describe an approach for characterizing selective binding between oppositely charged ionic functional groups under biologically
relevant conditions. Relative shifts in K-shell x-ray absorption spectra of aqueous cations and carboxylate anions indicate
the corresponding binding strengths via perturbations of carbonyl antibonding orbitals. XAS spectra measured for aqueous formate
and acetate solutions containing lithium, sodium, and potassium cations reveal monotonically stronger binding of the lighter
metals, supporting recent results from simulations and other experiments. The carbon K-edge spectra of the acetate carbonyl
feature centered near 290 eV clearly indicate a preferential interaction of sodium versus potassium, which was less apparent
with formate. These results are in accord with the Law of Matching Water Affinities, relating relative hydration strengths
of ions to their respective tendencies to form contact ion pairs. Density functional theory calculations of K-shell spectra
support the experimental findings.
Co-reporter:Jared D. Smith;Christopher D. Cappa;Kevin R. Wilson;Ronald C. Cohen;Phillip L. Geissler;
Proceedings of the National Academy of Sciences 2005 102(40) pp:14171-14174
Publication Date(Web):September 22, 2005
DOI:10.1073/pnas.0506899102
The unique chemical and physical properties of liquid water are a direct result of its highly directional hydrogen-bond (HB)
network structure and associated dynamics. However, despite intense experimental and theoretical scrutiny spanning more than
four decades, a coherent description of this HB network remains elusive. The essential question of whether continuum or multicomponent
(“intact,” “broken bond,” etc.) models best describe the HB interactions in liquid water has engendered particularly intense
discussion. Most notably, the temperature dependence of water's Raman spectrum has long been considered to be among the strongest
evidence for a multicomponent distribution. Using a combined experimental and theoretical approach, we show here that many
of the features of the Raman spectrum that are considered to be hallmarks of a multistate system, including the asymmetric
band profile, the isosbestic (temperature invariant) point, and van't Hoff behavior, actually result from a continuous distribution.
Furthermore, the excellent agreement between our newly remeasured Raman spectra and our model system further supports the
locally tetrahedral description of liquid water, which has recently been called into question [Wernet, P., et al. (2004) Science 304, 995-999].
Co-reporter:Jared D. Smith;Christopher D. Cappa;Kevin R. Wilson;Benjamin M. Messer;Ronald C. Cohen
Science 2004 Vol 306(5697) pp:851-853
Publication Date(Web):29 Oct 2004
DOI:10.1126/science.1102560
Abstract
A strong temperature dependence of oxygen K-edge x-ray absorption fine structure features was observed for supercooled and normal liquid water droplets prepared from the breakup of a liquid microjet. Analysis of the data over the temperature range 251 to 288 kelvin (–22° to +15°C) yields a value of 1.5 ± 0.5 kilocalories per mole for the average thermal energy required to effect an observable rearrangement between the fully coordinated (“ice-like”) and distorted (“broken-donor”) local hydrogen-bonding configurations responsible for the pre-edge and post-edge features, respectively. This energy equals the latent heat of melting of ice with hexagonal symmetry (ice Ih) and is consistent with the distribution of hydrogen bond strengths obtained for the “overstructured” ST2 model of water.
Co-reporter:Andrew M. Duffin, Alice H. England, Craig P. Schwartz, Janel S. Uejio, Gregory C. Dallinger, Orion Shih, David Prendergast and Richard J. Saykally
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 38) pp:NaN17083-17083
Publication Date(Web):2011/08/05
DOI:10.1039/C1CP21788G
Borohydride salts have been considered as good prospects for transportable hydrogen storage materials, with molecular hydrogen released via hydrolysis. We examine details of the hydration of sodium borohydride by the combination of X-ray absorption spectroscopy and first principles' theory. Compared to solid sodium borohydride, the aqueous sample exhibits an uncharacteristically narrow absorption feature that is shifted to lower energy, and ascribed to the formation of dihydrogen bonds between borohydride and water that weaken the boron–hydrogen covalent bonds. Water also acts to localize the highly excited molecular orbitals of borohydride, causing transitions to excited states with p character to become more intense and a sharp feature, uncharacteristic of tetrahedral molecules, to emerge. The simulations indicate that water preferentially associates with borohydride on the tetrahedral corners and edges.
Co-reporter:Kaitlin C. Duffey, Orion Shih, Nolan L. Wong, Walter S. Drisdell, Richard J. Saykally and Ronald C. Cohen
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 28) pp:NaN11639-11639
Publication Date(Web):2013/05/07
DOI:10.1039/C3CP51148K
The presence of organic surfactants in atmospheric aerosol may lead to a depression of cloud droplet growth and evaporation rates affecting the radiative properties and lifetime of clouds. Both the magnitude and mechanism of this effect, however, remain poorly constrained. We have used Raman thermometry measurements of freely evaporating micro-droplets to determine evaporation coefficients for several concentrations of acetic acid, which is ubiquitous in atmospheric aerosol and has been shown to adsorb strongly to the air–water interface. We find no suppression of the evaporation kinetics over the concentration range studied (1–5 M). The evaporation coefficient determined for 2 M acetic acid is 0.53 ± 0.12, indistinguishable from that of pure water (0.62 ± 0.09).
Co-reporter:Jacob W. Smith, Royce K. Lam, Alex T. Sheardy, Orion Shih, Anthony M. Rizzuto, Oleg Borodin, Stephen J. Harris, David Prendergast and Richard J. Saykally
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 43) pp:NaN23575-23575
Publication Date(Web):2014/08/20
DOI:10.1039/C4CP03240C
Since their introduction into the commercial marketplace in 1991, lithium ion batteries have become increasingly ubiquitous in portable technology. Nevertheless, improvements to existing battery technology are necessary to expand their utility for larger-scale applications, such as electric vehicles. Advances may be realized from improvements to the liquid electrolyte; however, current understanding of the liquid structure and properties remains incomplete. X-ray absorption spectroscopy of solutions of LiBF4 in propylene carbonate (PC), interpreted using first-principles electronic structure calculations within the eXcited electron and Core Hole (XCH) approximation, yields new insight into the solvation structure of the Li+ ion in this model electrolyte. By generating linear combinations of the computed spectra of Li+-associating and free PC molecules and comparing to the experimental spectrum, we find a Li+–solvent interaction number of 4.5. This result suggests that computational models of lithium ion battery electrolytes should move beyond tetrahedral coordination structures.
.jpg)













![Poly[imino[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/25300/25213-34-7.png)
![Poly[imino[(1S)-1-methyl-2-oxo-1,2-ethanediyl]]](http://img.cochemist.com/ccimg/25300/25213-34-7_b.png)