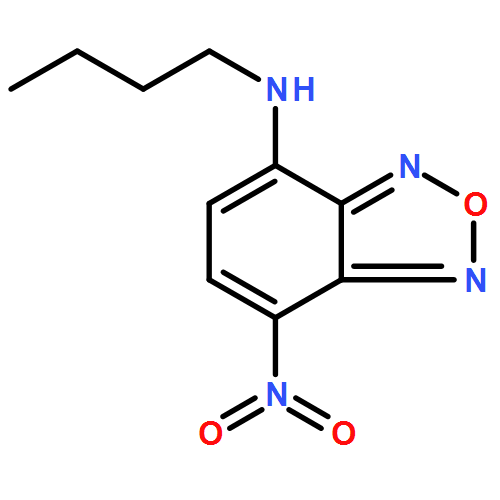
Co-reporter:Mariana Amaro, Hugo A. L. Filipe, J. P. Prates Ramalho, Martin Hof and Luís M. S. Loura
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 10) pp:7042-7054
Publication Date(Web):07 Dec 2015
DOI:10.1039/C5CP05238F
Nitrobenzoxadiazole (NBD)-labeled lipids are popular fluorescent membrane probes. However, the understanding of important aspects of the photophysics of NBD remains incomplete, including the observed shift in the emission spectrum of NBD-lipids to longer wavelengths following excitation at the red edge of the absorption spectrum (red-edge excitation shift or REES). REES of NBD-lipids in membrane environments has been previously interpreted as reflecting restricted mobility of solvent surrounding the fluorophore. However, this requires a large change in the dipole moment (Δμ) of NBD upon excitation. Previous calculations of the value of Δμ of NBD in the literature have been carried out using outdated semi-empirical methods, leading to conflicting values. Using up-to-date density functional theory methods, we recalculated the value of Δμ and verified that it is rather small (∼2 D). Fluorescence measurements confirmed that the value of REES is ∼16 nm for 1,2-dioleoyl-sn-glycero-3-phospho-L-serine-N-(NBD) (NBD-PS) in dioleoylphosphatidylcholine vesicles. However, the observed shift is independent of both the temperature and the presence of cholesterol and is therefore insensitive to the mobility and hydration of the membrane. Moreover, red-edge excitation leads to an increased contribution of the decay component with a shorter lifetime, whereas time-resolved emission spectra of NBD-PS displayed an atypical blue shift following excitation. This excludes restrictions to solvent relaxation as the cause of the measured REES and TRES of NBD, pointing instead to the heterogeneous transverse location of probes as the origin of these effects. The latter hypothesis was confirmed by molecular dynamics simulations, from which the calculated heterogeneity of the hydration and location of NBD correlated with the measured fluorescence lifetimes/REES. Globally, our combination of theoretical and experiment-based techniques has led to a considerably improved understanding of the photophysics of NBD and a reinterpretation of its REES in particular.
Co-reporter:João R. Robalo, J. P. Prates Ramalho, Daniel Huster and Luís M. S. Loura
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 35) pp:22736-22748
Publication Date(Web):31 Jul 2015
DOI:10.1039/C5CP03097H
Following a recent experimental investigation of the effect of the length of the alkyl side chain in a series of cholesterol analogues (Angew. Chem., Int. Ed., 2013, 52, 12848–12851), we report here an atomistic molecular dynamics characterization of the behaviour of methyl-branched side chain sterols (iso series) in POPC bilayers. The studied sterols included androstenol (i-C0-sterol) and cholesterol (i-C8-sterol), as well as four other derivatives (i-C5, i-C10, i-C12 and i-C14-sterol). For each sterol, both subtle local effects and more substantial differential alterations of membrane properties along the iso series were investigated. The location and orientation of the tetracyclic ring system is almost identical in all compounds. Among all the studied sterols, cholesterol is the sterol that presents the best matching with the hydrophobic length of POPC acyl chains, whereas longer-chained sterols interdigitate into the opposing membrane leaflet. In accordance with the experimental observations, a maximal ordering effect is observed for intermediate sterol chain length (i-C5, cholesterol, i-C10). Only for these sterols a preferential interaction with the saturated sn-1 chain of POPC (compared to the unsaturated sn-2 chain) was observed, but not for either shorter or longer-chained derivatives. This work highlights the importance of the sterol alkyl chain in the modulation of membrane properties and lateral organization in biological membranes.
Co-reporter:Hugo A. L. Filipe, Lennon S. Santos, J. P. Prates Ramalho, Maria João Moreno and Luís M. S. Loura
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 31) pp:20066-20079
Publication Date(Web):22 May 2015
DOI:10.1039/C5CP01596K
A complete homologous series of fluorescent phosphatidylethanolamines (diCnPE), labelled at the head group with a 7-nitrobenz-2-oxa-1,3-diazo-4-yl(NBD) fluorophore and inserted in 1-palmitoyl, 2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayers, was studied using atomistic molecular dynamics simulations. The longer-chained derivatives of NBD-diCnPE, with n = 14, 16, and 18, are commercially available, and widely used as fluorescent membrane probes. Properties such as location of atomic groups and acyl chain order parameters of both POPC and NBD-diCnPE, fluorophore orientation and hydrogen bonding, membrane electrostatic potential and lateral diffusion were calculated for all derivatives in the series. Most of these probes induce local disordering of POPC acyl chains, which is on the whole counterbalanced by ordering resulting from binding of sodium ions to lipid carbonyl/glycerol oxygen atoms. An exception is found for NBD-diC16PE, which displays optimal matching with POPC acyl chain length and induces a slight local ordering of phospholipid acyl chains. Compared to previously studied fatty amines, acyl chain-labelled phosphatidylcholines, and sterols bearing the same fluorescent tag, the chromophore in NBD-diCnPE locates in a similar region of the membrane (near the glycerol backbone/carbonyl region) but adopts a different orientation (with the NO2 group facing the interior of the bilayer). This modification leads to an inverted orientation of the P–N axis in the labelled lipid, which affects the interface properties, such as the membrane electrostatic potential and hydrogen bonding to lipid head group atoms. The implications of this study for the interpretation of the photophysical properties of NBD-diCnPE (complex fluorescence emission kinetics, differences with other NBD lipid probes) are discussed.
Co-reporter:Hugo A. L. Filipe, Maria João Moreno, Tomasz Róg, Ilpo Vattulainen, and Luís M. S. Loura
The Journal of Physical Chemistry B 2014 Volume 118(Issue 13) pp:3572-3581
Publication Date(Web):March 17, 2014
DOI:10.1021/jp501622d
One of the great challenges in membrane biophysics is to find a means to foster the transport of drugs across complex membrane structures. In this spirit, we elucidate methodological challenges associated with free energy computations of complex chainlike molecules across lipid membranes. As an appropriate standard molecule to this end, we consider 7-nitrobenz-2-oxa-1,3-diazol-4-yl-labeled fatty amine, NBD-Cn, which is here dealt with as a homologous series with varying chain lengths. We found the membrane–water interface region to be highly sensitive to details in free energy computations. Despite considerable simulation times, we observed substantial hysteresis, the cause being the small frequency of insertion/desorption events of the amphiphile’s alkyl chain in the membrane interface. The hysteresis was most pronounced when the amphiphile was pulled from water to the membrane and compromised the data that were not in line with experiments. The subtleties in umbrella sampling for computing distance along the transition path were also observed to be potential causes of artifacts. With the PGD (pull geometry distance) scheme, in which the distance from the molecule was computed to a reference plane determined by an average over all lipids in the membrane, we found marked deformations in membrane structure when the amphiphile was close to the membrane. The deformations were weaker with the PGC (pull geometry cylinder) method, where the reference plane is chosen based on lipids that are within a cylinder of radius 1.7 nm from the amphiphile. Importantly, the free energy results given by PGC were found to be qualitatively consistent with experimental data, while the PGD results were not. We conclude that with long amphiphiles there is reason for concern with regard to computations of their free energy profiles. The membrane–water interface is the region where the greatest care is warranted.
Co-reporter:João R. Robalo, J. P. Prates Ramalho, and Luís M. S. Loura
The Journal of Physical Chemistry B 2013 Volume 117(Issue 44) pp:13731-13742
Publication Date(Web):October 7, 2013
DOI:10.1021/jp406135a
Nitrobenzoxadiazole (NBD)-labeled sterols are commonly used as fluorescent cholesterol analogues in membrane biophysics. However, some experimental reports have questioned their ability to emulate the behavior of cholesterol in phospholipid bilayers. For the purpose of a detailed clarification of this matter, atomistic molecular dynamics simulations of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayers, containing either cholesterol or one of two fluorescent cholesterol analogues, 22-NBD-cholesterol or 25-NBD-cholesterol, were carried out. It is found that these sterol probes tend to adopt conformations in which their tail-labeled fluorophore is oriented toward the lipid/water interface, with a location similar to that observed in molecular dynamics simulations of other NBD probes. This implies that in these molecules the long sterol axis is no longer aligned with the membrane normal, and preferentially adopts orientations approximately parallel to the bilayer plane. In turn, these stretched conformations, together with NBD-POPC atomic interactions, lead to slowed-down lateral diffusion of both fluorescent sterols, compared to cholesterol. From computation of the deuterium order parameter and acyl chain tilts of POPC chains for varying POPC-sterol distance, it is observed that the local ordering effect of sterol is altered in both fluorescent derivatives. In agreement with reported experimental data, both fluorescent sterols are able to increase the order of POPC at 20 mol % concentration (as some molecules adopt an upright conformation, possibly related to formation of transbilayer aggregates), albeit to a smaller extent to that of cholesterol. Altogether, this study indicates that both 22- and 25-NBD-cholesterol are unable to mimic the most important features of cholesterol’s behavior in lipid bilayers.
Co-reporter:João R. Robalo, António M. T. Martins do Canto, A. J. Palace Carvalho, J. P. Prates Ramalho, and Luís M. S. Loura
The Journal of Physical Chemistry B 2013 Volume 117(Issue 19) pp:5806-5819
Publication Date(Web):April 18, 2013
DOI:10.1021/jp312026u
Molecular dynamics simulations of bilayer systems consisting of varying proportions of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), cholesterol (Chol), and intrinsically fluorescent Chol analogues dehydroergosterol (DHE) or cholestatrienol (CTL) were carried out to study in detail the extent to which these fluorescent probes mimic Chol’s behavior (location, orientation, dynamics) in membranes as well as their effect on host bilayer structure and dynamics (namely their ability to induce membrane ordering in comparison with Chol). Control properties of POPC and POPC/Chol bilayers agree well with published experimental and simulation work. Both probes and Chol share similar structural and dynamical properties within the bilayers. Additionally, the fluorescent sterols induce membrane ordering to a similar (slightly lower) extent to that of Chol. These findings combined demonstrate that the two studied fluorescent sterols are adequate analogues of Chol, and may be used with advantage over side-chain labeled sterols. The small structural differences between the three studied sterols are responsible for the slight variations in the calculated properties, with CTL presenting a more similar behavior to Chol (correlating with its larger structural similarity to Chol) compared to DHE.
Co-reporter:Ana Coutinho;Luís M. S. Loura;Manuel Prieto
Journal of Neurochemistry 2011 Volume 116( Issue 5) pp:696-701
Publication Date(Web):
DOI:10.1111/j.1471-4159.2010.07000.x
J. Neurochem. (2011) 116, 696–701.
Abstract
Acidic lipids are known to both catalyze amyloid fiber formation by amyloidogenic peptides/proteins and induce formation of ‘amyloid-like’ fibrils by non-amyloidogenic proteins. In this work, we describe the application of state-of-the-art time-resolved Förster resonance energy transfer methodologies to the characterization of the supramolecular structure of the aggregates formed by both a cationic peptide (hexalysyltryptophan) and a basic non-amyloidogenic protein (lysozyme) upon their interaction with negatively-charged fluid membranes (mixtures of zwitterionic phosphatidylcholine and anionic phosphatidylserine). It was concluded that both the peptide and protein induce the formation of multistacked lipid bilayers. Furthermore, upon using conditions that are described in the literature to cause the formation of amyloid-like fibers, lysozyme was found to induce the formation of a ‘pinched lamellar’ structure, with reduced interbilayer distance in the regions where there is bound protein, and increased interbilayer distance (stabilized by hydration repulsion) outside these areas. No significant lateral domains (lipid demixing) were induced in the membrane by either the cationic peptide or lysozyme.
Co-reporter:A.M.T. Martins do Canto, A.J. Palace Carvalho, J.P. Prates Ramalho, Luís M.S. Loura
Biophysical Chemistry 2011 Volume 159(2–3) pp:275-286
Publication Date(Web):December 2011
DOI:10.1016/j.bpc.2011.08.001
T-20 (also known as enfuvirtide) is a fusion inhibitor peptide known to have some effectiveness in the control of progression of HIV infection by inhibiting the fusion of the HIV envelope with the target cell membrane. Recent results indicate that T-20 is able to interact with membranes in the liquid disordered state but not with membranes in an ordered state, which could be linked to its effectiveness. A detailed molecular picture of the interaction of these molecules with membranes is still lacking. To this effect, extensive molecular dynamics simulations (100 ns) were carried out to investigate the interaction between T-20 and bilayers of 1-palmitoyl-2-oleoyl-phosphatidylcholine (POPC) and POPC/cholesterol (1:1). Membrane properties such as area/lipid, density profiles, order parameters and membrane thickness were studied. It was observed that T-20 has the ability to interact to different extents with both model membranes in this study and that peptide interaction with the bilayer surface has a local effect on membrane structure. The formation of hydrogen bonding between certain peptide residues and the POPC phosphate group was observed. However, T-20 showed a more limited extent of interaction with model membranes when compared with other, more efficient, peptides (such as T-1249). This effect is most notable in POPC/Chol membranes in which interaction is especially weak, owing to less peptide residues acting as H bond donors to POPC and virtually no H bonds being formed between T-20 and cholesterol. This lower ability to interact with membranes is probably correlated with its smaller inhibitory efficiency.Highlights► HIV fusion inhibitor T-20 (enfuvirtide) was studied by molecular dynamics simulations. ► The inhibitor peptide was simulated in both POPC and POPC/Cholesterol 1:1 bilayers. ► T-20 showed a more limited extent of interaction with model membranes than T-1249. ► Interaction with POPC/Cholesterol bilayers is especially weak, no penetration occurs. ► The weaker interaction with membranes correlates with a smaller inhibitory efficiency.
Co-reporter:Hugo A. L. Filipe, Maria João Moreno, and Luís M. S. Loura
The Journal of Physical Chemistry B 2011 Volume 115(Issue 33) pp:10109-10119
Publication Date(Web):July 12, 2011
DOI:10.1021/jp203532c
A complete homologous series of fluorescent 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD)-labeled fatty amines of varying alkyl chain length, NBD-Cn, inserted in 1-palmitoyl, 2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayers, was studied using atomistic molecular dynamics (MD) simulations. For all amphiphiles, the NBD fluorophore locates near the glycerol backbone/carbonyl region of POPC and establishes stable hydrogen bonding with POPC ester oxygen atoms. Small differences observed in the transverse location of the fluorophore correlate with other calculated parameters and with small discrepancies recently measured in the photophysical properties of the molecules. The longer-chained NBD-Cn amphiphiles show significant mass density near the bilayer midplane, and the chains of these derivatives interdigitate to some extent the opposite bilayer leaflet. This phenomenon leads to a slower lateral diffusion for the longer-chained derivatives (n > 12). Effects of these amphiphiles on the structure and dynamics of the host lipid were found to be relatively mild, in comparison with acyl-chain-labeled NBD probes. The molecular details obtained by this work allow the rationalization of the nonmonotonic behavior, recently obtained experimentally, for the photophysical parameters of the amphiphiles and the kinetic and thermodynamic parameters for their interaction with the POPC membranes.
Co-reporter:Luís M. S. Loura;Fábio Fernandes;Manuel Prieto
European Biophysics Journal 2010 Volume 39( Issue 4) pp:589-607
Publication Date(Web):2010 March
DOI:10.1007/s00249-009-0547-5
Lateral membrane heterogeneity, in the form of lipid rafts and microdomains, is currently implicated in cell processes including signal transduction, endocytosis, and cholesterol trafficking. Various biophysical techniques have been used to detect and characterize lateral membrane domains. Among these, Förster resonance energy transfer (FRET) has the crucial advantage of being sensitive to domain sizes smaller than 50-100 nm, below the resolution of optical microscopy but, apparently, similar to those of rafts in cell membranes. In the last decade, several formalisms for the analysis of FRET in heterogeneous membrane systems have been derived and applied to the study of microdomains. They are critically described and illustrated here.
Co-reporter:A.M.T. Martins do Canto, A.J. Palace Carvalho, J.P. Prates Ramalho, Luís M.S. Loura
Journal of Molecular Structure: THEOCHEM 2010 Volume 946(1–3) pp:119-124
Publication Date(Web):30 April 2010
DOI:10.1016/j.theochem.2009.12.010
Fusion of the HIV envelope with the target cell membrane is a critical step of viral entry into the target cell. Several peptides based on the C-region of HIV gp41 protein have been used in clinical trials as possible HIV fusion inhibitors, this way controlling the progression of the infection. Some of them interact strongly with ordered membranes, which could be linked to their effectiveness. In this way, extensive molecular dynamics simulations (100 ns) were carried out to investigate the structure, conformational behavior and dynamics of HIV fusion inhibitor peptide T-1249 in water and in the presence of bilayers of 1-palmitoyl-2-oleyl-phosphatidylcholine (POPC) and POPC–cholesterol (1:1). Peptide properties such as secondary structure, degree of membrane interaction and rotational and translational dynamics were analyzed. It was found that the peptide has a helical conformation in solution. It adsorbs readily to the membrane surface and remains tightly bound, with highly impeded rotational motion, keeping the helical structure. Whereas very limited penetration is observed in POPC, in the ordered POPC/cholesterol system the peptide just rests on top of the headgroup region of the bilayer, parallel to the surface. These results are rationalized in relation to recent experimental observations.
Co-reporter:Luís M. S. Loura;J. P. Prates Ramalho
Biophysical Reviews 2009 Volume 1( Issue 3) pp:
Publication Date(Web):2009 September
DOI:10.1007/s12551-009-0016-5
Fluorescence spectroscopy and microscopy have been used as tools to study membrane biophysics for decades now. Because phospholipids are non-fluorescent, the use of extrinsic membrane probes in this context is commonplace. Two major points of concern arise regarding this matter, namely the incomplete understanding of the probe behavior inside the bilayer and the perturbation of the latter resulting from probe incorporation. To this effect, molecular dynamics (MD) simulations, by providing detailed atomic-scale information, represent a valuable way to characterize the location and dynamics of bilayer-inserted membrane probes, as well as the magnitude of perturbation they induce on the host lipid structure, and several important classes of reporter molecules have been studied in recent years. This article reviews the state of the art of MD simulations of bilayer-inserted fluorescent probes, focusing on the information that has been obtained from previous studies and hinting at future perspectives in this rapidly emerging field.
Co-reporter:António M.T.M. do Canto, João R. Robalo, Patrícia D. Santos, Alfredo J. Palace Carvalho, J.P. Prates Ramalho, Luís M.S. Loura
Biochimica et Biophysica Acta (BBA) - Biomembranes (November 2016) Volume 1858(Issue 11) pp:
Publication Date(Web):November 2016
DOI:10.1016/j.bbamem.2016.07.013
•DPH and TMA-DPH were simulated in both pure POPC and POPC/cholesterol bilayers.•We provided insights on the behavior of TMA-DPH and its differences to that of DPH.•The effects of both probes on the bilayer properties are globally small.•TMA-DPH has a more superficial location than DPH, albeit by only ~ 0.3–0.4 nm.•Motions of TMA-DPH are hindered because of electrostatic interactions with lipids.Fluorescence spectroscopy and microscopy have been utilized as tools in membrane biophysics for decades now. Because phospholipids are non-fluorescent, the use of extrinsic membrane probes in this context is commonplace. Among the latter, 1,6-diphenylhexatriene (DPH) and its trimethylammonium derivative (TMA-DPH) have been extensively used. It is widely believed that, owing to its additional charged group, TMA-DPH is anchored at the lipid/water interface and reports on a bilayer region that is distinct from that of the hydrophobic DPH. In this study, we employ atomistic MD simulations to characterize the behavior of DPH and TMA-DPH in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and POPC/cholesterol (4:1) bilayers. We show that although the dynamics of TMA-DPH in these membranes is noticeably more hindered than that of DPH, the location of the average fluorophore of TMA-DPH is only ~ 3–4 Å more shallow than that of DPH. The hindrance observed in the translational and rotational motions of TMA-DPH compared to DPH is mainly not due to significant differences in depth, but to the favorable electrostatic interactions of the former with electronegative lipid atoms instead. By revealing detailed insights on the behavior of these two probes, our results are useful both in the interpretation of past work and in the planning of future experiments using them as membrane reporters.
Co-reporter:Luís M.S. Loura, Rodrigo F.M. de Almeida, Liana C. Silva, Manuel Prieto
Biochimica et Biophysica Acta (BBA) - Biomembranes (January 2009) Volume 1788(Issue 1) pp:
Publication Date(Web):January 2009
DOI:10.1016/j.bbamem.2008.10.012
The application of Förster Resonance Energy Transfer (FRET) to the detection and characterization of phase separation in lipid bilayers (both in model systems and in cell membranes) is reviewed. Models describing the rate and efficiency of FRET for both uniform probe distribution and phase separation, and recently reported methods for detection of membrane heterogeneity and determination of phase boundaries, probe partition coefficients and domain size, are presented and critically discussed. Selected recent applications of FRET to one-phase lipid systems, gel/fluid phase separation, liquid ordered/liquid disordered phase separation (lipid rafts), complex systems containing ceramide and cell membranes are presented to illustrate the wealth of information that can be inferred from carefully designed FRET studies of membrane domains.
Co-reporter:Luís M.S. Loura, Fábio Fernandes, A.C. Fernandes, J.P. Prates Ramalho
Biochimica et Biophysica Acta (BBA) - Biomembranes (February 2008) Volume 1778(Issue 2) pp:
Publication Date(Web):February 2008
DOI:10.1016/j.bbamem.2007.10.022
We present a combined theoretical (molecular dynamics, MD) and experimental (differential scanning calorimetry, DSC) study of the effect of 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD) acyl chain-labeled fluorescent phospholipid analogs (C6-NBD-PC and C12-NBD-PC) on 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) bilayers. DSC measurements reveal that < 1 mol% of NBD-PC causes elimination of the pre-transition and a large loss of cooperativity of the main transition of DPPC. Labeling with C6-NBD-PC or C12-NBD-PC shifts the main transition temperature to lower or higher values, respectively. Following our recent report on the location and dynamics of these probes (BBA 1768 (2007) 467–478) in fluid phase DPPC, we present a detailed analysis of 100-ns MD simulations of systems containing either C6-NBD-PC or C12-NBD-PC, focused on their influence on several properties of the host bilayer. Whereas most monitored parameters are not severely affected for 1.6 mol% of probe, for the higher concentration studied (6.2 mol%) important differences are evident. In agreement with published reports, we observed that the average area per phospholipid molecule increases, whereas DPPC acyl chain order parameters decrease. Moreover, we predict that incorporation of NBD-PC should increase the electrostatic potential across the bilayer and, especially for C12-NBD-PC, slow lateral diffusion of DPPC molecules and rotational mobility of DPPC acyl chains.
Co-reporter:João R. Robalo, J. P. Prates Ramalho, Daniel Huster and Luís M. S. Loura
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 35) pp:NaN22748-22748
Publication Date(Web):2015/07/31
DOI:10.1039/C5CP03097H
Following a recent experimental investigation of the effect of the length of the alkyl side chain in a series of cholesterol analogues (Angew. Chem., Int. Ed., 2013, 52, 12848–12851), we report here an atomistic molecular dynamics characterization of the behaviour of methyl-branched side chain sterols (iso series) in POPC bilayers. The studied sterols included androstenol (i-C0-sterol) and cholesterol (i-C8-sterol), as well as four other derivatives (i-C5, i-C10, i-C12 and i-C14-sterol). For each sterol, both subtle local effects and more substantial differential alterations of membrane properties along the iso series were investigated. The location and orientation of the tetracyclic ring system is almost identical in all compounds. Among all the studied sterols, cholesterol is the sterol that presents the best matching with the hydrophobic length of POPC acyl chains, whereas longer-chained sterols interdigitate into the opposing membrane leaflet. In accordance with the experimental observations, a maximal ordering effect is observed for intermediate sterol chain length (i-C5, cholesterol, i-C10). Only for these sterols a preferential interaction with the saturated sn-1 chain of POPC (compared to the unsaturated sn-2 chain) was observed, but not for either shorter or longer-chained derivatives. This work highlights the importance of the sterol alkyl chain in the modulation of membrane properties and lateral organization in biological membranes.
Co-reporter:Mariana Amaro, Hugo A. L. Filipe, J. P. Prates Ramalho, Martin Hof and Luís M. S. Loura
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 10) pp:NaN7054-7054
Publication Date(Web):2015/12/07
DOI:10.1039/C5CP05238F
Nitrobenzoxadiazole (NBD)-labeled lipids are popular fluorescent membrane probes. However, the understanding of important aspects of the photophysics of NBD remains incomplete, including the observed shift in the emission spectrum of NBD-lipids to longer wavelengths following excitation at the red edge of the absorption spectrum (red-edge excitation shift or REES). REES of NBD-lipids in membrane environments has been previously interpreted as reflecting restricted mobility of solvent surrounding the fluorophore. However, this requires a large change in the dipole moment (Δμ) of NBD upon excitation. Previous calculations of the value of Δμ of NBD in the literature have been carried out using outdated semi-empirical methods, leading to conflicting values. Using up-to-date density functional theory methods, we recalculated the value of Δμ and verified that it is rather small (∼2 D). Fluorescence measurements confirmed that the value of REES is ∼16 nm for 1,2-dioleoyl-sn-glycero-3-phospho-L-serine-N-(NBD) (NBD-PS) in dioleoylphosphatidylcholine vesicles. However, the observed shift is independent of both the temperature and the presence of cholesterol and is therefore insensitive to the mobility and hydration of the membrane. Moreover, red-edge excitation leads to an increased contribution of the decay component with a shorter lifetime, whereas time-resolved emission spectra of NBD-PS displayed an atypical blue shift following excitation. This excludes restrictions to solvent relaxation as the cause of the measured REES and TRES of NBD, pointing instead to the heterogeneous transverse location of probes as the origin of these effects. The latter hypothesis was confirmed by molecular dynamics simulations, from which the calculated heterogeneity of the hydration and location of NBD correlated with the measured fluorescence lifetimes/REES. Globally, our combination of theoretical and experiment-based techniques has led to a considerably improved understanding of the photophysics of NBD and a reinterpretation of its REES in particular.
Co-reporter:Hugo A. L. Filipe, Lennon S. Santos, J. P. Prates Ramalho, Maria João Moreno and Luís M. S. Loura
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 31) pp:NaN20079-20079
Publication Date(Web):2015/05/22
DOI:10.1039/C5CP01596K
A complete homologous series of fluorescent phosphatidylethanolamines (diCnPE), labelled at the head group with a 7-nitrobenz-2-oxa-1,3-diazo-4-yl(NBD) fluorophore and inserted in 1-palmitoyl, 2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayers, was studied using atomistic molecular dynamics simulations. The longer-chained derivatives of NBD-diCnPE, with n = 14, 16, and 18, are commercially available, and widely used as fluorescent membrane probes. Properties such as location of atomic groups and acyl chain order parameters of both POPC and NBD-diCnPE, fluorophore orientation and hydrogen bonding, membrane electrostatic potential and lateral diffusion were calculated for all derivatives in the series. Most of these probes induce local disordering of POPC acyl chains, which is on the whole counterbalanced by ordering resulting from binding of sodium ions to lipid carbonyl/glycerol oxygen atoms. An exception is found for NBD-diC16PE, which displays optimal matching with POPC acyl chain length and induces a slight local ordering of phospholipid acyl chains. Compared to previously studied fatty amines, acyl chain-labelled phosphatidylcholines, and sterols bearing the same fluorescent tag, the chromophore in NBD-diCnPE locates in a similar region of the membrane (near the glycerol backbone/carbonyl region) but adopts a different orientation (with the NO2 group facing the interior of the bilayer). This modification leads to an inverted orientation of the P–N axis in the labelled lipid, which affects the interface properties, such as the membrane electrostatic potential and hydrogen bonding to lipid head group atoms. The implications of this study for the interpretation of the photophysical properties of NBD-diCnPE (complex fluorescence emission kinetics, differences with other NBD lipid probes) are discussed.