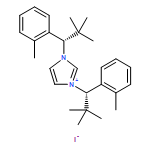
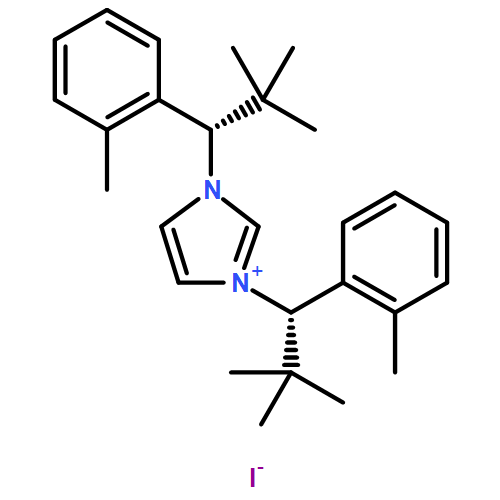
Invited for the cover of this issue is the group of E. Peter Kündig at the University of Geneva. The cover illustrates the regiodivergent reaction taken by two enantiomers when subjected to a chiral Pd(N-heterocyclic carbene) catalyst. The combination of a divergent reaction with an enantioselective CH functionalization makes for fascinating chemistry. Read the full text of the article at 10.1002/chem.201403985.
Two bulky, chiral, monodentate N-heterocyclic carbene ligands were applied to palladium-catalyzed asymmetric CH arylation to incorporate C(sp3)H bond activation. Racemic mixtures of the carbamate starting materials underwent regiodivergent reactions to afford different trans-2,3-substituted indolines. Although this CArCalkyl coupling requires high temperatures (140–160 °C), chiral induction is high. This regiodivergent reaction, when carried out with enantiopure starting materials, can lead to single structurally different enantiopure products, depending on the catalyst chirality. The CH activation at a tertiary center was realized only in the case of a cyclopropyl group. No CH activation takes place alpha to a tertiary center. A detailed DFT study is included and analyses of methyl versus methylene versus methine CH activation is used to rationalize experimentally observed regio- and enantioselectivities.
No abstract is available for this article.
The highly tuned, one-point binding cationic cyclopentadienyl-iron and -ruthenium complexes 1 and 2 that incorporate chiral bidentate pentafluoroaryl-phosphinite ligands selectively coordinate and activate α,β-unsaturated carbonyl compounds towards asymmetric catalytic cycloaddition reactions with diaryl nitrones. The reaction gives isoxazolidine products in good yields, with complete endo selectivity and high enantioselectivity. The products are obtained as a mixture of regioisomers in ratios varying from 96:4 to 15:85. The regioselectivity correlates directly with the electronic properties of the nitrone. This is shown by the experimental and computational data.
Bring on the big cats: New, C2-symmetric bulky N-heterocyclic carbene ligands bring major improvements in the palladium-catalyzed asymmetric intramolecular α-arylation of amides to give oxindoles (see picture, dba=trans,trans-dibenzylideneacetone), which are formed in high yield and excellent enantiomeric purity.
The complex [Ru(Cp)(R,R-BIPHOP-F)(acetone)][SbF6], (R,R)-1 a, was used as catalyst for asymmetric Diels–Alder reactions between dienes (cyclopentadiene, methylcyclopentadiene, isoprene, 2,3-dimethylbutadiene) and α,β-unsaturated ketones (methyl vinyl ketone (MVK), ethyl vinyl ketone, divinyl ketone, α-bromovinyl methyl ketone and α-chlorovinyl methyl ketone). The cycloaddition products were obtained in yields of 50–90 % and with enantioselectivities up to 96 % ee. Ethyl vinyl ketone, divinyl ketone and the halogenated vinyl ketones worked best and their reactions with acyclic dienes consistently provided products with >90 % ee. α-Chlorovinyl methyl ketone performed better than α-bromovinyl methyl ketone. The reaction also provided a [4.3.1]bicyclic ring system in 95 % ee through an intramolecular cycloaddition reaction. Crystal structure determinations of [Ru(Cp)((S,S)-BIPHOP-F)(mvk)][SbF6], (S,S)-1 b, and [Ru(Cp)((R,R)-Me4BIPHOP-F)(acrolein)][SbF6], (R,R)-2 b, provided the basis for a rationalization of the asymmetric induction.
Gezielte Spaltung: In einer Untersuchung zur asymmetrischen Hydrogenolyse einer Arylkohlenstoff-Brom-Bindung in einem Naphthalinchromtricarbonyl-Komplex erwies sich ein neuer sperriger Phosphoramiditligand als ideal, um das Produkt hoch enantiomerenangereichert zu erhalten (siehe Schema).
Lithiation/electrophile trapping reactions were carried out with the highly enantiomerically enriched complex [Cr(5-bromonaphthalene)(CO)3]. Electrophile quenching with ClPPh2, PhCHO, and (Me3SiO)2 afforded the enantiomerically enriched (>97 % ee) planar chiral 5-substituted naphthalene complexes with PPh2, CH(Ph)OH, and OH substituents, respectively. Very mild Pd-catalyzed Suzuki–Miyaura cross-coupling reactions were developed and applied to the highly labile [Cr(5-bromonaphthalene)(CO)3] to give nine new planar chiral aryl-, heteroaryl-, alkynyl-, and alkenylnaphthalene chromium complexes with high enantiomeric purity. The efficient ambient-temperature coupling reactions with borinates prepared in situ were also applied to a number of chlorobenzene complexes and to aryl and vinyl halides.
An efficient dearomatization process of [Cr(arene)(CO)3] complexes initiated by a nucleophilic acetaldehyde equivalent is detailed. It generates in a one-pot reaction three CC bonds and two stereogenic centers. This process allowed a rapid assembly of a cis-decalin ring system incorporating a homoannular diene unit in just two steps starting from aromatic precursors (Scheme 2). The method was applied to the total synthesis of the eudesmane-type marine furanosesquiterpene (±)-15-acetoxytubipofuran (2). Two routes were successfully used to synthesize the γ-lactone precursor of the furan ring. The key step in the first approach was a Pd-catalyzed allylic substitution (Scheme 3), while in the second approach, an Eschenmoser–Claisen rearrangement was highly successful (Scheme 4). The Pd-catalyzed allylic substitution could be directed to give either the (normal) product with overall retention as major diastereoisomer or the unusual product with inversion of configuration (see Table). For the synthesis of the (−)-enantiomer (R,R)-2 of 15-acetoxytubipofuran, an enantioselective dearomatization in the presence of a chiral diether ligand was implemented (Scheme 7), while the (+)-enantiomer (S,S)-2 was obtained via a diastereoselective dearomatization of an arene-bound chiral imine auxiliary (Scheme 8). Chiroptical data suggest that a revision of the previously assigned absolute configuration of the natural product is required.
[Ru(η5-C5H5)(CH3CN)3][PF6] (1), a versatile precursor to catalysts containing the CpRu+ complex fragment, can be obtained in high yield via Cp/naphthalene exchange in ruthenocene. This new route via the complex [RuCp(naphthalene)][PF6] (4) avoids both highly toxic thallium reagents and the need for a photoreactor.
Keine CO-Insertion erfolgt bei der ersten Mo(CO)3-vermittelten Dearomatisierungs-Reaktionssequenz (siehe Schema). Anders als bei der entsprechenden Reaktion mit Cr(CO)3 lässt sich der intermediäre Allylkomplex isolieren.
No CO insertion occurs in the first Mo(CO)3-mediated dearomatization reaction sequence (shown). In contrast to the analogous Cr(CO)3 reaction, the intermediate allyl complex can be isolated.
Chiral iron and ruthenium Lewis acids: analogies and differences between the catalysts and the role of the anion in catalytic Diels-Alder reaction. In short: Fe catalysts are faster but Ru analogues are more stable and can be recovered quantitatively. Rational ligand design is shown to result in a large increase in chiral induction.
The new chiral bidentate (phosphinoaryl)benzoxazine ligands 2 were applied in asymmetric catalysis. Rhodium and copper complexes catalyzed the hydrosilylation of acetophenone and [4+2] cycloadditions with moderate enantioselectivity. Iridium complexes were used to hyrogenate di-, tri-, and tetrasubstituted alkenes, giving products with moderate to high enantiomer excesses. Enantioselective allylic substitution and Heck reactions catalyzed by [(phosphinoaryl)benzoxazine]palladium complexes occurred with high enantioselectivities. The results were similar to those obtained with the corresponding dihydro(phosphinoaryl)oxazole ligands. Comparison of the structures of (diphenylallyl)(benzoxazine)palladium and (diphenylallyl)(dihydrooxazole)palladium complexes showed that the coordination geometries and the chiral environments of the metal centers are very similar, which explains why the enantioselectivities induced by the two ligand classes are in the same range.
Das Indenyldach über dem katalytischen Zentrum der chiralen Lewis-Säure [(Indenyl)Ru((S,S)-biphop-F)]+ (siehe Bild) führt nicht nur zu hohen Enantioselektivitäten bei Diels-Alder-Reaktionen von Enalen mit Cyclopentadien, sondern beeinflusst zudem die endo/exo-Selektivität, wobei das exo-Produkt selbst mit Acrolein bevorzugt gebildet wird.
The indenyl roof over the chiral Lewis acid catalyst site of [(indenyl)Ru((S,S)-biphop-F)]+ (see picture) gives rise not only to high enantioselectivities in Diels–Alder reactions between enals and cyclopentadiene but also affects the endo/exo diastereoselectivity. The exo product is formed preferentially even with acrolein.
Efficient routes to α-tert-butyl- and α-iso-propyl-ortho-hydroxybenzylamines 1a and 1b are described. Highly enantioenriched 1a and 1b were obtained by resolution of the methoxy derivatives 2 by recrystallization of the salts formed with mandelic acid followed by Lewis acid mediated demethylation. The chiral 1,3-amino alcohol 1a has also been obtained in an asymmetric synthesis with the key step a diastereoselective alkylation of the imine obtained by condensation of o-anisaldehyde with phenyl glycinol. The absolute stereochemistry of these 1,3-aminophenols was determined by CD spectroscopy of the salicylideneamines 12 and by an X-ray structure analysis of the salt formed between (R)-mandelic acid and (S)-α-tert-butyl-ortho-methoxybenzylamine ((S)-2a). Chirality 12:529–539, 2000. © 2000 Wiley-Liss, Inc.
Der einfache Zugang, die Stabilität in Lösung bei Raumtemperatur, die hohe Enantioselektivität in Diels-Alder-Reaktionen, die Rückgewinnbarkeit und die hohen Reaktivitätsunterschiede bei Variation des Gegenions kennzeichnen den neuartigen Ru-Lewis-Säure-Katalysator [CpRu((S,S)-biphop-F)]+ (biphop-F=(C6F5)2POCH2(Ph)CH2(Ph)OP(C6F5)2). Die Struktur des Komplexes 1 (L = Methacrolein; Y = SbF6) deutet auf die kooperative Bindung des Dienophils sowohl durch die Lewis-Säure als auch durch das Anion.
Ease of generation, stablity in solution at ambient temperature, high enantioselectivity in Diels–Alder reactions, efficient catalyst recovery, and large rate differences on variation of the anion are all characteristics of the new Ru Lewis acid [CpRu((S,S)-biphop-F)]+ (biphop-F=(C6F5)2POCH2(Ph)CH2(Ph)OP(C6F5)2). The structure of complex 1 (L=methacrolein, Y=SbF6) provides evidence for a cooperative binding of the dienophile by both the Lewis acid and the anion.
Leichter verdrängen läßt sich der Benzolligand in 1 im Vergleich zum Methylacrylatligand, die beide bei Raumtemperatur labil sind. Damit sind Aren-Substitutions- sowie -Austauschreaktionen möglich, die vermutlich durch eine η2η4-Umlagerung eingeleitet werden. Der Komplex 1 ist daher eine unter milden Bedingungen zugängliche Vorstufe für die in hohem Maße ungesättigten Fragmente [Cr(CO)2] (siehe Schema) und [Cr(CO)2(η2-acrylat)].
The surprising answer to the question “Are the structural characteristics of the product of the nucleophilic addition to complex 1 those of a cyclohexadienyl complex (2 a) or those of a diene aza-enolate complex (2 b)?” is: both. This is indicated by the X-ray structure determination of the complex with R=naphthyl. An unprecedented endo-hydride abstraction concludes the sequence that provides a new route to 1,2-substituted planar chiral [(η6-arene)Cr(CO)3] compounds.
Displacement of the benzene ligand in 1 surprisingly occurs more readily than that of the methyl acrylate ligand. This paves the way for 1 to undergo arene exchange and arene substitution reactions, which may be triggered by a η2η4 haptotropic rearrangement of the acrylate. Complex 1 is thus a mild precursor of the highly unsaturated fragments [Cr(CO)2] (see scheme) and [Cr(CO)2(η2-acrylate)].