Co-reporter:Robert P. Rambo;Michael W. W. Adams;Jack H. Freed;Ashley J. Pratt;John A. Tainer;Kevin N. Dyer;Farris L. Poole, II;Brian R. Crane;Gregory E. Merz;W. Andrew Lancaster;Peter P. Borbat;David S. Shin
PNAS 2014 Volume 111 (Issue 43 ) pp:E4568-E4576
Publication Date(Web):2014-10-28
DOI:10.1073/pnas.1308531111
Protein framework alterations in heritable Cu, Zn superoxide dismutase (SOD) mutants cause misassembly and aggregation in
cells affected by the motor neuron disease ALS. However, the mechanistic relationship between superoxide dismutase 1 (SOD1) mutations and human disease is controversial, with many hypotheses postulated for the propensity of specific SOD mutants
to cause ALS. Here, we experimentally identify distinguishing attributes of ALS mutant SOD proteins that correlate with clinical
severity by applying solution biophysical techniques to six ALS mutants at human SOD hotspot glycine 93. A small-angle X-ray
scattering (SAXS) assay and other structural methods assessed aggregation propensity by defining the size and shape of fibrillar
SOD aggregates after mild biochemical perturbations. Inductively coupled plasma MS quantified metal ion binding stoichiometry,
and pulsed dipolar ESR spectroscopy evaluated the Cu2+ binding site and defined cross-dimer copper–copper distance distributions. Importantly, we find that copper deficiency in
these mutants promotes aggregation in a manner strikingly consistent with their clinical severities. G93 mutants seem to properly
incorporate metal ions under physiological conditions when assisted by the copper chaperone but release copper under destabilizing
conditions more readily than the WT enzyme. Altered intradimer flexibility in ALS mutants may cause differential metal retention
and promote distinct aggregation trends observed for mutant proteins in vitro and in ALS patients. Combined biophysical and
structural results test and link copper retention to the framework destabilization hypothesis as a unifying general mechanism
for both SOD aggregation and ALS disease progression, with implications for disease severity and therapeutic intervention
strategies.
Co-reporter:Andrew S. Arvai;Monika Heilmann;Ashley J. Pratt;Sharon M. Kelly;Katherine J. Baxter;Andrew O’Hara;John M. Christie;Michael Hothorn;Brian O. Smith;Kenichi Hitomi;Gareth I. Jenkins
Science 2012 Volume 335(Issue 6075) pp:1492-1496
Publication Date(Web):23 Mar 2012
DOI:10.1126/science.1218091
Co-reporter:Noriyuki Nishimura;Kenichi Hitomi;Andrew S. Arvai;Robert P. Rambo;Chiharu Hitomi;Sean R. Cutler;Julian I. Schroeder
Science 2009 Volume 326(Issue 5958) pp:1373-1379
Publication Date(Web):04 Dec 2009
DOI:10.1126/science.1181829
Co-reporter:Luciano DiTacchio;Andrew S. Arvai;Sang-Tae Kim;Takeshi Todo;Satchidananda Panda;Shigenori Iwai;Kenichi Hitomi;Junpei Yamamoto;John A. Tainer
PNAS 2009 Volume 106 (Issue 17 ) pp:6962-6967
Publication Date(Web):2009-04-28
DOI:10.1073/pnas.0809180106
Homologous flavoproteins from the photolyase (PHR)/cryptochrome (CRY) family use the FAD cofactor in PHRs to catalyze DNA
repair and in CRYs to tune the circadian clock and control development. To help address how PHR/CRY members achieve these
diverse functions, we determined the crystallographic structure of Arabidopsis thaliana (6-4) PHR (UVR3), which is strikingly (>65%) similar in sequence to human circadian clock CRYs. The structure reveals a substrate-binding
cavity specific for the UV-induced DNA lesion, (6-4) photoproduct, and cofactor binding sites different from those of bacterial
PHRs and consistent with distinct mechanisms for activities and regulation. Mutational analyses were combined with this prototypic
structure for the (6-4) PHR/clock CRY cluster to identify structural and functional motifs: phosphate-binding and Pro-Lys-Leu
protrusion motifs constricting access to the substrate-binding cavity above FAD, sulfur loop near the external end of the
Trp electron-transfer pathway, and previously undefined C-terminal helix. Our results provide a detailed, unified framework
for investigations of (6-4) PHRs and the mammalian CRYs. Conservation of key residues and motifs controlling FAD access and
activities suggests that regulation of FAD redox properties and radical stability is essential not only for (6-4) photoproduct
DNA repair, but also for circadian clock-regulating CRY functions. The structural and functional results reported here elucidate
archetypal relationships within this flavoprotein family and suggest how PHRs and CRYs use local residue and cofactor tuning,
rather than larger structural modifications, to achieve their diverse functions encompassing DNA repair, plant growth and
development, and circadian clock regulation.
Co-reporter:
Nature Structural and Molecular Biology 2003 10(12) pp:1064-1073
Publication Date(Web):02 November 2003
DOI:10.1038/nsb1007
Sulfur metabolism depends on the iron-containing porphinoid siroheme. In Salmonella enterica, the S-adenosyl-L-methionine (SAM)-dependent bismethyltransferase, dehydrogenase and ferrochelatase, CysG, synthesizes siroheme from uroporphyrinogen III (uro'gen III). The reactions mediated by CysG encompass two branchpoint intermediates in tetrapyrrole biosynthesis, diverting flux first from protoporphyrin IX biosynthesis and then from cobalamin (vitamin B12) biosynthesis. We determined the first structure of this multifunctional siroheme synthase by X-ray crystallography. CysG is a homodimeric gene fusion product containing two structurally independent modules: a bismethyltransferase and a dual-function dehydrogenase-chelatase. The methyltransferase active site is a deep groove with a hydrophobic patch surrounded by hydrogen bond donors. This asymmetric arrangement of amino acids may be important in directing substrate binding. Notably, our structure shows that CysG is a phosphoprotein. From mutational analysis of the post-translationally modified serine, we suggest a conserved role for phosphorylation in inhibiting dehydrogenase activity and modulating metabolic flux between siroheme and cobalamin pathways.
Co-reporter:
Nature Structural and Molecular Biology 2003 10(8) pp:663-668
Publication Date(Web):20 July 2003
DOI:10.1038/nsb958
Protein photoreceptors use small-molecule cofactors called chromophores to detect light. Only under the influence of the receptors' active sites do these chromophores adopt spectral and photochemical properties that suit the receptors' functional requirements. This protein-induced change in chromophore properties is called photochemical tuning and is a prime example for the general—but poorly understood—process of chemical tuning through which proteins shape the reactivity of their active-site groups. Here we report the 0.82-Å resolution X-ray structure of the bacterial light receptor photoactive yellow protein (PYP). The unusually precise structure reveals deviations from expected molecular geometries and anisotropic atomic displacements in the PYP active site. Our analysis of these deviations points directly to the intramolecular forces and active-site dynamics that tune the properties of PYP's chromophore to absorb blue light, suppress fluorescence, and favor the required light-driven double-bond isomerization.
Co-reporter:David P. Barondeau;Carey J. Kassmann;John A. Tainer;Christopher D. Putnam
PNAS 2003 Volume 100 (Issue 21 ) pp:12111-12116
Publication Date(Web):2003-10-14
DOI:10.1073/pnas.2133463100
Green fluorescent protein has revolutionized cell labeling and molecular tagging, yet the driving force and mechanism for
its spontaneous fluorophore synthesis are not established. Here we discover mutations that substantially slow the rate but
not the yield of this posttranslational modification, determine structures of the trapped precyclization intermediate and
oxidized postcyclization states, and identify unanticipated features critical to chromophore maturation. The protein architecture
contains a dramatic ≈80° bend in the central helix, which focuses distortions at G67 to promote ring formation from amino
acids S65, Y66, and G67. Significantly, these distortions eliminate potential helical hydrogen bonds that would otherwise
have to be broken at an energetic cost during peptide cyclization and force the G67 nitrogen and S65 carbonyl oxygen atoms
within van der Waals contact in preparation for covalent bond formation. Further, we determine that under aerobic, but not
anaerobic, conditions the Gly-Gly-Gly chromophore sequence cyclizes and incorporates an oxygen atom. These results lead directly
to a conjugation-trapping mechanism, in which a thermodynamically unfavorable cyclization reaction is coupled to an electronic
conjugation trapping step, to drive chromophore maturation. Moreover, we propose primarily electrostatic roles for the R96
and E222 side chains in chromophore formation and suggest that the T62 carbonyl oxygen is the base that initiates the dehydration
reaction. Our molecular mechanism provides the basis for understanding and eventually controlling chromophore creation.
Co-reporter:Ulrich K. Genick,
S. Michael Soltis,
Peter Kuhn,
Ilona L. Canestrelli
and
Elizabeth D. Getzoff
Nature 1998 392(6672) pp:206
Publication Date(Web):
DOI:10.1038/32462
Protein photosensors from all kingdoms of life1,2 use bound organic molecules, known as chromophores, to detect light. A specific double bond within each chromophore is isomerized by light, triggering slower changes in the protein as a whole. The initial movements of the chromophore, which can occur in femtoseconds, are tightly constrained by the surrounding protein, making it difficult to see how isomerization can occur, be recognized, and be appropriately converted into a protein-wide structural change and biological signal. Here we report how this dilemma is resolved in the photoactive yellow protein (PYP). We trapped a key early intermediate in the light cycle of PYP at temperatures below -100 °C, and determined its structure at better than 1 Å resolution. The 4-hydroxycinnamoyl chromophore3,4 isomerizes by flipping its thioester linkage with the protein, thus avoiding collisions resulting from large-scale movement of its aromatic ring during the initial light reaction. A protein-to-chromophore hydrogen bond that is present in both the preceding dark state5 and the subsequent signalling state6 of the photosensor breaks, forcing one of the hydrogen-bonding partners into a hydrophobic pocket. The isomerized bond is distorted into a conformation resembling that in the transition state. The resultant stored energy is used to drive the PYP light cycle. These results suggest a model for phototransduction, with implications for bacteriorhodopsin7,8, photoactive proteins1,2, PAS domains9, and signalling proteins.
.jpg)
![Pentanoic acid,5-[[4-[[hydroxy[(4-nitrophenyl)amino]phosphinyl]methyl]phenyl]amino]-5-oxo-](http://img.cochemist.com/ccimg/135700/135690-33-4.png)
![Pentanoic acid,5-[[4-[[hydroxy[(4-nitrophenyl)amino]phosphinyl]methyl]phenyl]amino]-5-oxo-](http://img.cochemist.com/ccimg/135700/135690-33-4_b.png)


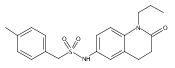
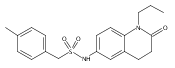


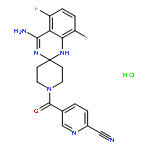
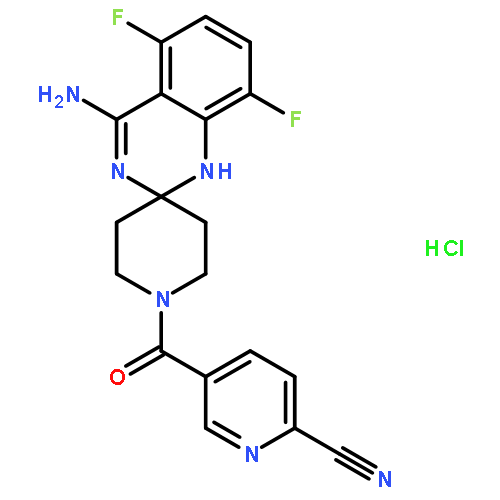


![Pentanoic acid,5-[[4-[2-[(4-nitrophenyl)amino]-2-oxoethyl]phenyl]amino]-5-oxo-](http://img.cochemist.com/ccimg/117800/117746-37-9.png)
![Pentanoic acid,5-[[4-[2-[(4-nitrophenyl)amino]-2-oxoethyl]phenyl]amino]-5-oxo-](http://img.cochemist.com/ccimg/117800/117746-37-9_b.png)






![L-Ornithine,N5-[(hydroxyamino)iminomethyl]-](http://img.cochemist.com/ccimg/53100/53054-07-2.png)
![L-Ornithine,N5-[(hydroxyamino)iminomethyl]-](http://img.cochemist.com/ccimg/53100/53054-07-2_b.png)


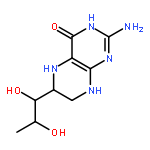






![4(3H)-Pteridinone,2-amino-6-[(1R,2S)-1,2-dihydroxypropyl]-7,8-dihydro-](http://img.cochemist.com/ccimg/6800/6779-87-9.png)
![4(3H)-Pteridinone,2-amino-6-[(1R,2S)-1,2-dihydroxypropyl]-7,8-dihydro-](http://img.cochemist.com/ccimg/6800/6779-87-9_b.png)

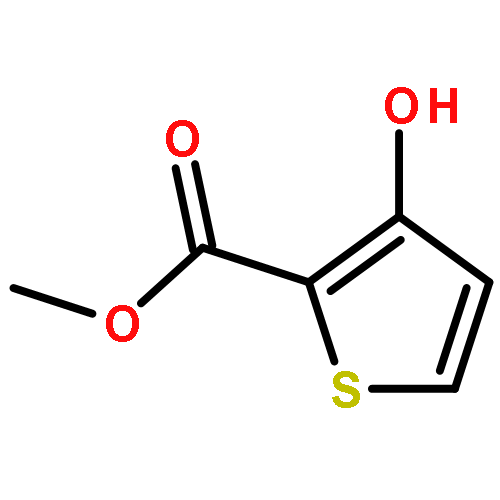


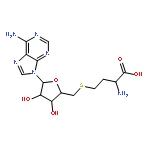
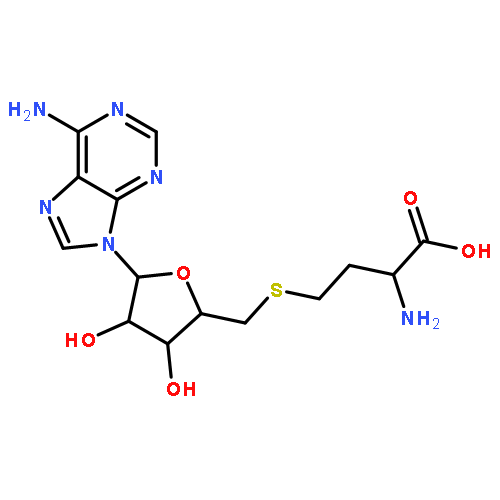


















![Ferrate(8-),[(12S,13S,17S,18S)-2,8,13,18-tetrakis(carboxymethyl)-12,13,17,18-tetrahydro-13,18-dimethyl-21H,22H-porphine-3,7,12,17-tetrapropanoato(10-)-kN21,kN22,kN23,kN24]-, hydrogen (1:8), (SP-4-2)-](http://img.cochemist.com/ccimg/52600/52553-42-1.png)
![Ferrate(8-),[(12S,13S,17S,18S)-2,8,13,18-tetrakis(carboxymethyl)-12,13,17,18-tetrahydro-13,18-dimethyl-21H,22H-porphine-3,7,12,17-tetrapropanoato(10-)-kN21,kN22,kN23,kN24]-, hydrogen (1:8), (SP-4-2)-](http://img.cochemist.com/ccimg/52600/52553-42-1_b.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)







