Co-reporter:Florence P. Varodayan, Diego Correia, Dean Kirson, Sophia Khom, Christopher S. Oleata, George Luu, Paul Schweitzer, Marisa Roberto
Neuropharmacology 2017 Volume 125(Volume 125) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.neuropharm.2017.08.009
•CRF acts primarily via CRF1 at central amygdala glutamatergic synapses.•CRF decreases evoked glutamate transmission at central amygdala synapses.•CRF increases spontaneous glutamate release at central amygdala synapses.•CRF affects glutamate transmission similarly in naïve and ethanol-dependent rats.Corticotropin-releasing factor (CRF) signaling in the central nucleus of the amygdala (CeA) is hypothesized to drive the development of alcohol dependence, as it regulates ethanol intake and several anxiogenic behaviors linked to withdrawal. Excitatory glutamatergic neurotransmission contributes to alcohol reinforcement, tolerance and dependence. Therefore, in this study we used in vitro slice electrophysiology to investigate the effects of CRF and its receptor subtype (CRF1 and CRF2) antagonists on both evoked and spontaneous action potential-independent glutamatergic transmission in the CeA of naive and ethanol-dependent Sprague-Dawley rats. We found that CRF (25–200 nM) concentration-dependently diminished evoked compound excitatory postsynaptic potentials (EPSPs), but increased miniature excitatory postsynaptic current (mEPSC) frequencies similarly in CeA neurons of both naïve and ethanol-dependent rats, indicating reduced evoked glutamatergic responses and enhanced vesicular glutamate release, respectively. This CRF-induced vesicular glutamate release was prevented by the CRF1/2 antagonist (Astressin B) and the CRF1 antagonist (R121919), but not by the CRF2 antagonist (Astressin 2B). Similarly, CRF's effects on evoked glutamatergic responses were completely blocked by CRF1 antagonism, but only slightly decreased in the presence of the CRF2 antagonist. Moreover, CRF1 antagonism reveals a tonic facilitation of vesicular glutamate, whereas the CRF2 antagonism revealed a tonic inhibition of vesicular glutamate release. Collectively our data show that CRF primarily acts at presynaptic CRF1 to produce opposite effects on CeA evoked and spontaneous glutamate release and that the CRF system modulates CeA glutamatergic synapses throughout the development of alcohol dependence.
Co-reporter:Marisa Roberto, Florence P. Varodayan
Neuropharmacology 2017 Volume 122(Volume 122) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.neuropharm.2017.01.013
•Chronic alcohol exposure produces neuroadaptation at GABA and glutamate synapses.•The main effects are altered GABAAR/NMDAR expression, composition and/or function.•It is hypothesized that these adaptations play a role in addictive drinking behavior.Alcohol acts on numerous cellular and molecular targets to regulate neuronal communication within the brain. Chronic alcohol exposure and acute withdrawal generate prominent neuroadaptations at synapses, including compensatory effects on the expression, localization and function of synaptic proteins, channels and receptors.The present article reviews the literature describing the synaptic effects of chronic alcohol exposure and their relevance for synaptic transmission in the central nervous system. This review is not meant to be comprehensive, but rather to highlight the effects that have been observed most consistently and that are thought to contribute to the development of alcohol dependence and the negative aspects of withdrawal. Specifically, we will focus on the major excitatory and inhibitory neurotransmitters in the brain, glutamate and GABA, respectively, and how their neuroadaptations after chronic alcohol exposure contributes to alcohol reinforcement, dependence and withdrawal.This article is part of the Special Issue entitled “Alcoholism”.
Co-reporter:Luis A. Natividad, Matthew W. Buczynski, Melissa A. Herman, Dean Kirson, ... Loren H. Parsons
Biological Psychiatry 2017 Volume 82, Issue 7(Volume 82, Issue 7) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.biopsych.2017.01.005
BackgroundCorticotropin-releasing factor (CRF) mediates anxiogenic responses by activating CRF type 1 (CRF1) receptors in limbic brain regions. Anxiety is further modulated by the endogenous cannabinoid (eCB) system that attenuates the synaptic effects of stress. In the amygdala, acute stress activates the enzymatic clearance of the eCB N-arachidonoylethanolamine via fatty acid amide hydrolase (FAAH), although it is unclear whether chronic dysregulation of CRF systems induces maladaptive changes in amygdalar eCB signaling. Here, we used genetically selected Marchigian Sardinian P (msP) rats carrying an innate overexpression of CRF1 receptors to study the role of constitutive upregulation in CRF systems on amygdalar eCB function and persistent anxiety-like effects.MethodsWe applied behavioral, pharmacological, and biochemical methods to broadly characterize anxiety-like behaviors and amygdalar eCB clearance enzymes in msP versus nonselected Wistar rats. Subsequent studies examined the influence of dysregulated CRF and FAAH systems in altering excitatory transmission in the central amygdala (CeA).ResultsmsPs display an anxious phenotype accompanied by elevations in amygdalar FAAH activity and reduced dialysate N-arachidonoylethanolamine levels in the CeA. Elevations in CRF–CRF1 signaling dysregulate FAAH activity, and this genotypic difference is normalized with pharmacological blockade of CRF1 receptors. msPs also exhibit elevated baseline glutamatergic transmission in the CeA, and dysregulated CRF–FAAH facilitates stress-induced increases in glutamatergic activity. Treatment with an FAAH inhibitor relieves sensitized glutamatergic responses in msPs and attenuates the anxiety-like phenotype.ConclusionsPathological anxiety and stress hypersensitivity are driven by constitutive increases in CRF1 signaling that dysregulate N-arachidonoylethanolamine signaling mechanisms and reduce neuronal inhibitory control of CeA glutamatergic synapses.
Co-reporter:Marsida Kallupi, Florence P Varodayan, Christopher S Oleata, Diego Correia, George Luu and Marisa Roberto
Neuropsychopharmacology 2014 39(5) pp:1081-1092
Publication Date(Web):November 27, 2013
DOI:10.1038/npp.2013.308
The central nucleus of the amygdala (CeA) mediates several addiction-related processes and nociceptin/orphanin FQ (nociceptin) regulates ethanol intake and anxiety-like behaviors. Glutamatergic synapses, in the CeA and throughout the brain, are very sensitive to ethanol and contribute to alcohol reinforcement, tolerance, and dependence. Previously, we reported that in the rat CeA, acute and chronic ethanol exposures significantly decrease glutamate transmission by both pre- and postsynaptic actions. In this study, using electrophysiological techniques in an in vitro CeA slice preparation, we investigated the effects of nociceptin on glutamatergic transmission and its interaction with acute ethanol in naive and ethanol-dependent rats. We found that nociceptin (100–1000 nM) diminished basal-evoked compound glutamatergic receptor-mediated excitatory postsynaptic potentials (EPSPs) and spontaneous and miniature EPSCs (s/mEPSCs) by mainly decreasing glutamate release in the CeA of naive rats. Notably, nociceptin blocked the inhibition induced by acute ethanol (44 mM) and ethanol blocked the nociceptin-induced inhibition of evoked EPSPs in CeA neurons of naive rats. In neurons from chronic ethanol-treated (ethanol-dependent) rats, the nociceptin-induced inhibition of evoked EPSP amplitude was not significantly different from that in naive rats. Application of [Nphe1]Nociceptin(1–13)NH2, a nociceptin receptor (NOP) antagonist, revealed tonic inhibitory activity of NOP on evoked CeA glutamatergic transmission only in ethanol-dependent rats. The antagonist also blocked nociceptin-induced decreases in glutamatergic responses, but did not affect ethanol-induced decreases in evoked EPSP amplitude. Taken together, these studies implicate a potential role for the nociceptin system in regulating glutamatergic transmission and a complex interaction with ethanol at CeA glutamatergic synapses.
Co-reporter:Maureen T Cruz, Melissa A Herman, Dawn M Cote, Andrey E Ryabinin and Marisa Roberto
Neuropsychopharmacology 2013 38(2) pp:364-375
Publication Date(Web):September 12, 2012
DOI:10.1038/npp.2012.190
The neural circuitry that processes natural rewards converges with that engaged by addictive drugs. Because of this common neurocircuitry, drugs of abuse have been able to engage the hedonic mechanisms normally associated with the processing of natural rewards. Ghrelin is an orexigenic peptide that stimulates food intake by activating GHS-R1A receptors in the hypothalamus. However, ghrelin also activates GHS-R1A receptors on extrahypothalamic targets that mediate alcohol reward. The central nucleus of the amygdala (CeA) has a critical role in regulating ethanol consumption and the response to ethanol withdrawal. We previously demonstrated that rat CeA GABAergic transmission is enhanced by acute and chronic ethanol treatment. Here, we used quantitative RT-PCR (qRT-PCR) to detect Ghsr mRNA in the CeA and performed electrophysiological recordings to measure ghrelin effects on GABA transmission in this brain region. Furthermore, we examined whether acute or chronic ethanol treatment would alter these electrophysiological effects. Our qRT-PCR studies show the presence of Ghsr mRNA in the CeA. In naive animals, superfusion of ghrelin increased the amplitude of evoked inhibitory postsynaptic potentials (IPSPs) and the frequency of miniature inhibitory postsynaptic currents (mIPSCs). Coapplication of ethanol further increased the ghrelin-induced enhancement of IPSP amplitude, but to a lesser extent than ethanol alone. When applied alone, ethanol significantly increased IPSP amplitude, but this effect was attenuated by the application of ghrelin. In neurons from chronic ethanol-treated (CET) animals, the magnitude of ghrelin-induced increases in IPSP amplitude was not significantly different from that in naive animals, but the ethanol-induced increase in amplitude was abolished. Superfusion of the GHS-R1A antagonists D-Lys3-GHRP-6 and JMV 3002 decreased evoked IPSP and mIPSC frequency, revealing tonic ghrelin activity in the CeA. D-Lys3-GHRP-6 and JMV 3002 also blocked ghrelin-induced increases in GABAergic responses. Furthermore, D-Lys3-GHRP-6 did not affect ethanol-induced increases in IPSP amplitude. These studies implicate a potential role for the ghrelin system in regulating GABAergic transmission and a complex interaction with ethanol at CeA GABAergic synapses.
Co-reporter:Michal Bajo;George R. Siggins;Robert Messing;Maureen T. Cruz
PNAS 2008 Volume 105 (Issue 24 ) pp:8410-8415
Publication Date(Web):2008-06-17
DOI:10.1073/pnas.0802302105
In the central amygdala (CeA), ethanol acts via corticotrophin-releasing factor (CRF) type 1 receptors to enhance GABA release.
Amygdala CRF mediates anxiety associated with stress and drug dependence, and it regulates ethanol intake. Because mutant
mice that lack PKCε exhibit reduced anxiety-like behavior and alcohol consumption, we investigated whether PKCε lies downstream
of CRF1 receptors in the CeA. Compared with PKCε+/+ CeA neurons, PKCε−/− neurons showed increased GABAergic tone due to enhanced GABA release. CRF and ethanol stimulated GABA release in the PKCε+/+ CeA, but not in the PKCε−/− CeA. A PKCε-specific inhibitor blocked both CRF- and ethanol-induced GABA release in the PKCε+/+ CeA, confirming findings in the PKCε−/− CeA. These results identify a PKCε signaling pathway in the CeA that is activated by CRF1 receptor stimulation, mediates GABA release at nerve terminals, and regulates anxiety and alcohol consumption.
Co-reporter:Marisa Roberto, Michal Bajo, Elena Crawford, Samuel G Madamba and George R Siggins
Neuropsychopharmacology 2006 31(5) pp:988-996
Publication Date(Web):July 27, 2005
DOI:10.1038/sj.npp.1300840
We recently reported that chronic ethanol treatment (CET) and early withdrawal (2–8 h) altered glutamatergic transmission at both pre- and postsynaptic sites in central nucleus of the amygdala (CeA). Acute ethanol (44 mM) inhibited the NMDA receptor (NMDAR)-mediated EPSCs (NMDA-EPSCs) more in CeA neurons from CET rats than from naïve rats and also decreased paired-pulse facilitation (PPF) of NMDA-EPSCs only in CET rats. To determine whether these CET effects persisted after prolonged withdrawal, we recorded intracellularly in rat CeA slices and measured mRNA and protein expression of CeA NMDAR subunits from CET rats and those withdrawn from ethanol for 1 or 2 weeks. At 1 week withdrawal, acute ethanol decreased evoked NMDA-EPSC amplitudes and NMDA currents induced by exogenous NMDA (20%) equally to that in naïve rats, indicating that CET effects on postsynaptic mechanisms reversed 1 week after CET cessation. However, acute ethanol still decreased PPF of NMDA-EPSCs, indicating that the acute ethanol-induced increase in glutamate release in CeA seen in CET rats was still present at this time. CET also significantly increased mRNA levels of NR1 and NR2B NMDAR subunits compared to control rats. At 1 week withdrawal, mRNA levels for NR1 and NR2B subunits were significantly decreased. These changes reversed at 2 weeks withdrawal. In Western blots, a significant increase in protein for all three subunits occurred in CeA from CET rats, but not after 1 and 2 weeks of withdrawal. These data indicate that CET induces reversible neuroadaptations in synaptic function, gene expression, and protein composition of NMDAR at CeA synapses.
Co-reporter:Marisa Roberto;George R. Siggins
PNAS 2006 Volume 103 (Issue 25 ) pp:9715-9720
Publication Date(Web):2006-06-20
DOI:10.1073/pnas.0601899103
Behavioral studies show that the GABAergic system in the central amygdala (CeA) nucleus has a complex role in the reinforcing
effects effects of ethanol and the anxiogenic response to ethanol withdrawal. Opioid peptides and nociceptin/orphanin FQ (nociceptin)
within the CeA are implicated also in regulating voluntary ethanol consumption and ethanol relapse. Recently, we reported
that basal GABAergic transmission was increased in ethanol-dependent rats, and that acute ethanol increases GABAA receptor-mediated inhibitory postsynaptic currents (IPSCs) in CeA neurons from both naïve and ethanol-dependent rats to the
same extent, suggesting lack of tolerance for the acute effect of ethanol. Here, we investigated the effect of nociceptin
on IPSCs in CeA neurons and its interaction with ethanol effects on these GABA synapses. We found that nociceptin moderately
decreased IPSC amplitudes, acting mostly presynaptically as it increased paired-pulse facilitation ratio of IPSCs and decreased
miniature IPSC frequencies (but not amplitudes). Nociceptin also prevented the ethanol-induced augmentation of IPSCs in CeA
of naïve rats. Interestingly, in CeA of ethanol-dependent rats, the nociceptin-induced inhibition of IPSCs was increased,
indicating an enhanced sensitivity to nociceptin. Nociceptin also blocked the ethanol-induced augmentation of IPSCs in ethanol-dependent
rats. Our data suggest that nociceptin has a role in regulating the GABAergic system and opposing the effect elicited by ethanol.
Thus, nociceptin may represent a therapeutic target for alleviating alcohol dependence.
Co-reporter:Marisa Roberto, George F. Koob
Alcohol (November 2009) Volume 43(Issue 7) pp:489-490
Publication Date(Web):November 2009
DOI:10.1016/j.alcohol.2009.10.008
Co-reporter:Melissa A. Herman, Florence P. Varodayan, Christopher S. Oleata, George Luu, Dean Kirson, Markus Heilig, Roberto Ciccocioppo, Marisa Roberto
Neuropharmacology (March 2016) Volume 102() pp:21-31
Publication Date(Web):1 March 2016
DOI:10.1016/j.neuropharm.2015.10.027
•CeA glutamate release is elevated in msP rats compared to Wistar rats.•EtOH produces divergent effects on glutamate release in msP and Wistar rats.•The increase in CeA glutamate release with EtOH is less in msP than in Wistar rats.•CRF produces divergent effects on glutamate release in Wistar rats.•In CeA neurons of msP rats CRF only increases glutamate release.The CRF system of the central nucleus of the amygdala (CeA) is important for the processing of anxiety, stress, and effects of acute and chronic ethanol. We previously reported that ethanol decreases evoked glutamate transmission in the CeA of Sprague Dawley rats and that ethanol dependence alters glutamate release in the CeA. Here, we examined the effects of ethanol, CRF and a CRF1 receptor antagonist on spontaneous and evoked glutamatergic transmission in CeA neurons from Wistar and Marchigian Sardinian Preferring (msP) rats, a rodent line genetically selected for excessive alcohol drinking and characterized by heightened activity of the CRF1 system. Basal spontaneous and evoked glutamate transmission in CeA neurons from msP rats was increased compared to Wistar rats. Ethanol had divergent effects, either increasing or decreasing spontaneous glutamate release in the CeA of Wistar rats. This bidirectional effect was retained in msP rats, but the magnitude of the ethanol-induced increase in glutamate release was significantly smaller. The inhibitory effect of ethanol on evoked glutamatergic transmission was similar in both strains. CRF also either increased or decreased spontaneous glutamate release in CeA neurons of Wistar rats, however, in msP rats CRF only increased glutamate release. The inhibitory effect of CRF on evoked glutamatergic transmission was also lost in neurons from msP rats. A CRF1 antagonist produced only minor effects on spontaneous glutamate transmission, which were consistent across strains, and no effects on evoked glutamate transmission. These results demonstrate that the genetically altered CRF system of msP rats results in alterations in spontaneous and stimulated glutamate signaling in the CeA that may contribute to both the anxiety and drinking behavioral phenotypes.
Co-reporter:Marisa Roberto, Thomas L. Kash, Patrick J. Mulholland, Vincent N. Marty, Nicholas W. Gilpin, Brendan M. Walker
Alcohol (June 2012) Volume 46(Issue 4) pp:301-302
Publication Date(Web):June 2012
DOI:10.1016/j.alcohol.2012.04.001
Co-reporter:Marisa Roberto, Florence Varodayan
Alcohol (December 2015) Volume 49(Issue 8) pp:
Publication Date(Web):1 December 2015
DOI:10.1016/j.alcohol.2015.11.007
Co-reporter:Maureen T. Cruz, Melissa A. Herman, Marsida Kallupi, Marisa Roberto
Biological Psychiatry (15 April 2012) Volume 71(Issue 8) pp:666-676
Publication Date(Web):15 April 2012
DOI:10.1016/j.biopsych.2011.10.032
Co-reporter:Marsida Kallupi, Sunmee Wee, Scott Edwards, Timothy W. Whitfield Jr., Christopher S. Oleata, George Luu, Brooke E. Schmeichel, George F. Koob, Marisa Roberto
Biological Psychiatry (1 October 2013) Volume 74(Issue 7) pp:520-528
Publication Date(Web):1 October 2013
DOI:10.1016/j.biopsych.2013.04.028
.jpg)
![3-(6-(Dimethylamino)-4-methylpyridin-3-yl)-2,5-dimethyl-N,N-dipropylpyrazolo[1,5-a]pyrimidin-7-amine](http://img.cochemist.com/ccimg/195100/195055-03-9.png)
![3-(6-(Dimethylamino)-4-methylpyridin-3-yl)-2,5-dimethyl-N,N-dipropylpyrazolo[1,5-a]pyrimidin-7-amine](http://img.cochemist.com/ccimg/195100/195055-03-9_b.png)
![4,8:11,15-Dimethano-20H-bisbenzofuro[2,3-a:3',2'-i]dipyrido[4,3-b:3',4'-h]carbazole-1,8a,10a,18-tetrol,7,12-bis(cyclopropylmethyl)-5,6,7,8,9,10,11,12,13,14,19a,20b-dodecahydro-,(4bS,8R,8aS,10aS,11R,14aS,19aR,20bR)-](http://img.cochemist.com/ccimg/105700/105618-26-6.png)
![4,8:11,15-Dimethano-20H-bisbenzofuro[2,3-a:3',2'-i]dipyrido[4,3-b:3',4'-h]carbazole-1,8a,10a,18-tetrol,7,12-bis(cyclopropylmethyl)-5,6,7,8,9,10,11,12,13,14,19a,20b-dodecahydro-,(4bS,8R,8aS,10aS,11R,14aS,19aR,20bR)-](http://img.cochemist.com/ccimg/105700/105618-26-6_b.png)






![5,8,11,14-Eicosatetraenamide,N-[(1R)-2-hydroxy-1-methylethyl]-, (5Z,8Z,11Z,14Z)-](http://img.cochemist.com/ccimg/157200/157182-49-5.png)
![5,8,11,14-Eicosatetraenamide,N-[(1R)-2-hydroxy-1-methylethyl]-, (5Z,8Z,11Z,14Z)-](http://img.cochemist.com/ccimg/157200/157182-49-5_b.png)


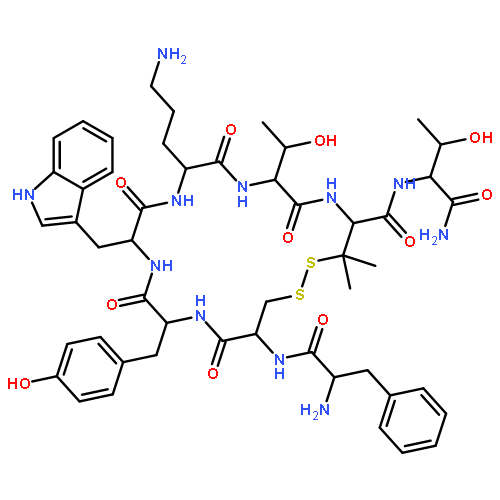
![acetic acid; (2S)-2-amino-N-[(1R)-1-[[[(1S)-1-(2-hydroxyethylcarbamoyl)-2-phenyl-ethyl]-methyl-carbamoyl]methylcarbamoyl]ethyl]-3-(4-hydroxyphenyl)propanamide](http://img.cochemist.com/ccimg/101000/100929-53-1.png)
![acetic acid; (2S)-2-amino-N-[(1R)-1-[[[(1S)-1-(2-hydroxyethylcarbamoyl)-2-phenyl-ethyl]-methyl-carbamoyl]methylcarbamoyl]ethyl]-3-(4-hydroxyphenyl)propanamide](http://img.cochemist.com/ccimg/101000/100929-53-1_b.png)






![DAMGO;[D-ALA2,NME-PHE4,GLY-OL5]-ENKEPHALIN](http://img.cochemist.com/ccimg/78200/78123-71-4.png)
![DAMGO;[D-ALA2,NME-PHE4,GLY-OL5]-ENKEPHALIN](http://img.cochemist.com/ccimg/78200/78123-71-4_b.png)


![Isoxazolo[5,4-c]pyridin-3(2H)-one,4,5,6,7-tetrahydro-](http://img.cochemist.com/ccimg/64700/64603-91-4.png)
![Isoxazolo[5,4-c]pyridin-3(2H)-one,4,5,6,7-tetrahydro-](http://img.cochemist.com/ccimg/64700/64603-91-4_b.png)










![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)
![Phosphinic acid,[3-[[1-(3,4-dichlorophenyl)ethyl]amino]-2-hydroxypropyl](phenylmethyl)- (9CI)](http://img.cochemist.com/ccimg/148100/148056-42-2.png)
![Phosphinic acid,[3-[[1-(3,4-dichlorophenyl)ethyl]amino]-2-hydroxypropyl](phenylmethyl)- (9CI)](http://img.cochemist.com/ccimg/148100/148056-42-2_b.png)





![Benzo[f]quinoxaline-7-sulfonamide,1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-](http://img.cochemist.com/ccimg/118900/118876-58-7.png)
![Benzo[f]quinoxaline-7-sulfonamide,1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-](http://img.cochemist.com/ccimg/118900/118876-58-7_b.png)