Co-reporter:Riad Mansour, Phillip S. Nejman, Laurence J. Taylor, Brian A. Chalmers, Fernando Rey, Abdelkader Bengueddach, J. Derek Woollins, Alexandra M.Z. Slawin, Petr Kilian
Polyhedron 2017 Volume 133(Volume 133) pp:
Publication Date(Web):5 September 2017
DOI:10.1016/j.poly.2017.05.050
A synthetic route to multigram quantities of 1-adamantyltrimethylphosphonium iodide is reported. The synthesis starts from the commercially available precursor 1-adamantyl bromide and was optimised with respect to yield and ease of purification. The title compound is of interest to zeolite chemists as a potent organic structure-directing agent. Full spectroscopic characterisation data of all isolated intermediates and single crystal X-ray diffraction data of AdP(O)Cl2, [AdPMe2H]I and [AdPMe3]I (Ad = 1-adamantyl) are reported.A synthetic route to multigram quantities of 1-adamantyltrimethylphosphonium iodide is reported, together with single crystal X-ray diffraction data of AdP(O)Cl2, [AdPMe2H]I and [AdPMe3]I (Ad = 1-adamantyl).Download high-res image (76KB)Download full-size image
Co-reporter:Brian A. Chalmers, Christina B. E. Meigh, Phillip S. Nejman, Michael Bühl, Tomáš Lébl, J. Derek Woollins, Alexandra M. Z. Slawin, and Petr Kilian
Inorganic Chemistry 2016 Volume 55(Issue 14) pp:7117-7125
Publication Date(Web):June 24, 2016
DOI:10.1021/acs.inorgchem.6b01079
Tris(acenaphthyl)- and bis(acenaphthyl)-substituted pnictogens (iPr2P-Ace)3E (2–4) (E = As, Sb, or Bi; Ace = acenaphthene-5,6-diyl) and (iPr2P-Ace)2EPh (5 and 6) (E = As or Sb) were synthesized and fully characterized by multinuclear nuclear magnetic resonance (NMR), high-resolution mass spectrometry, elemental analysis, and single-crystal X-ray diffraction. The molecules adopt propeller-like geometries with the restricted rotational freedom of the sterically encumbered iPr2P-Ace groups resulting in distinct NMR features. In the tris(acenaphthyl) species (2–4), the phosphorus atoms are isochronous in the 31P{1H} NMR spectra, and the rotation of the three acenaphthyl moieties around the E–Cipso bond is locked. On the other hand, the bis(acenaphthyl) species show a fluxional behavior, resulting in an AX to A2 spin system transition in the 31P{1H} variable-temperature NMR spectra. This allowed elucidation of remarkable through-space couplings (8TSJPP) of 11.5 Hz (for 5) and 25.8 Hz (for 6) at low temperatures. In addition, detailed line shape analysis of the thermodynamic parameters of the restricted rotation of the “propeller blades” in 5 was performed in the intermediate temperature region and also at coalescence. The lone pairs on the pnictogen atoms in 2–6 are oriented such that they form a bowl-shaped area that is somehow buried within the molecule.
Co-reporter:Laurence J. Taylor, Brian A. Surgenor, Piotr Wawrzyniak, Matthew J. Ray, David B. Cordes, Alexandra M. Z. Slawin and Petr Kilian
Dalton Transactions 2016 vol. 45(Issue 5) pp:1976-1986
Publication Date(Web):21 Aug 2015
DOI:10.1039/C5DT02539G
Bis(borane) adducts Acenap(PiPr2·BH3)(PRH·BH3) (Acenap = acenaphthene-5,6-diyl; 4a, R = Ph; 4b, R = ferrocenyl, Fc; 4c, R = H) were synthesised by the reaction of excess H3B·SMe2 with either phosphino-phosphonium salts [Acenap(PiPr2)(PR)]+Cl− (1a, R = Ph; 1b, R = Fc), or bis(phosphine) Acenap(PiPr2)(PH2) (3). Bis(borane) adducts 4a–c were found to undergo dihydrogen elimination at room temperature, this spontaneous catalyst-free phosphine-borane dehydrocoupling yields BH2 bridged species Acenap(PiPr2)(μ-BH2)(PR·BH3) (5a, R = Ph; 5b, R = Fc; 5c, R = H). Thermolysis of 5c results in loss of the terminal borane moiety to afford Acenap(PiPr2)(μ-BH2)(PH) (14). Single crystal X-ray structures of 3, 4b and 5a–c are reported.
Co-reporter:Laurence J. Taylor;Michael Bühl;Piotr Wawrzyniak;Brian A. Chalmers;J. Derek Woollins;Alexra M. Z. Slawin;Amy L. Fuller
European Journal of Inorganic Chemistry 2016 Volume 2016( Issue 5) pp:659-666
Publication Date(Web):
DOI:10.1002/ejic.201500948
Abstract
Treatment of Acenap(PiPr2)(EH2) (Acenap = acenaphthene-5,6-diyl; 1a, E = As; 1b, E = P) with Ph3C·BF4 resulted in hydride abstraction to give [Acenap(PiPr2)(EH)][BF4] (2a, E = As; 2b, E = P). These represent the first structurally characterised phosphino/arsino-phosphonium salts with secondary arsine/phosphine groups, as well as the first example of a Lewis base stabilised primary arsenium cation. Compounds 2a and 2b were deprotonated with NaH to afford low coordinate species Acenap(PiPr2)(E) (3a, E = As; 3b, E = P). This provides an alternative and practical synthetic pathway to the phosphanylidene-σ4-phosphorane 3b and provides mechanistic insight into the formation of arsanylidene-σ4-phosphorane 3a, indirectly supporting the hypothesis that the previously reported dehydrogenation of 1a occurs via an ionic mechanism.
Co-reporter:Brian A. Surgenor, Laurence J. Taylor, Andreas Nordheider, Alexandra M. Z. Slawin, Kasun S. Athukorala Arachchige, J. Derek Woollins and Petr Kilian
RSC Advances 2016 vol. 6(Issue 7) pp:5973-5976
Publication Date(Web):08 Jan 2016
DOI:10.1039/C5RA27594F
A new synthetic route to FcPH2 and FcPCl2 (Fc = ferrocenyl) is presented. This method avoids the challenging monolithiation of ferrocene, as well as any tedious purification steps. All reactions are high yielding and easily conducted on a relatively large scale, using economical and commercially available synthetic precursors.
Co-reporter:Brian A. Chalmers; Michael Bühl;Dr. Kasun S. AthukoralaArachchige; Alexra M. Z. Slawin ;Dr. Petr Kilian
Chemistry - A European Journal 2015 Volume 21( Issue 20) pp:7520-7531
Publication Date(Web):
DOI:10.1002/chem.201500281
Abstract
A series of phosphine–stibine and phosphine–stiborane peri-substituted acenaphthenes containing all permutations of pentavalent groups SbClnPh4–n (5–9), as well as trivalent groups SbCl2, Sb(R)Cl, and SbPh2 (2–4, R=Ph, Mes), were synthesised and fully characterised by single crystal diffraction and multinuclear NMR spectroscopy. In addition, the bonding in these species was studied by DFT computational methods. The P–Sb dative interactions in both series range from strongly bonding to non-bonding as the Lewis acidity of the Sb acceptor is decreased. In the pentavalent antimony series, a significant change in the P–Sb distance is observed between SbClPh3 and SbCl2Ph2 derivatives 6 and 7, respectively, consistent with a change from a bonding to a non-bonding interaction in response to relatively small modification in Lewis acidity of the acceptor. In the SbIII series, two geometric forms are observed. The P–Sb bond length in the SbCl2 derivative 2 is as expected for a normal (rather than a dative) bond. Rather unexpectedly, the phosphine–stiborane complexes 5–9 represent the first examples of the σ4Pσ6Sb structural motif.
Co-reporter:Brian A. Chalmers ; Michael Bühl ; Kasun S. Athukorala Arachchige ; Alexandra M. Z. Slawin
Journal of the American Chemical Society 2014 Volume 136(Issue 17) pp:6247-6250
Publication Date(Web):April 15, 2014
DOI:10.1021/ja502625z
A proximate Lewis basic group facilitates the mild dehydrogenative P–As intramolecular coupling in the phosphine-arsine peri-substituted acenaphthene 3, affording thermally and hydrolytically stable arsanylidine-phosphorane 4 with a sterically accessible two-coordinate arsenic atom. The formation of 4 is thermoneutral due to the dehydrogenation being concerted with the donor coordination. Reaction of 4 with a limited amount of oxygen reveals arsinidene-like reactivity via formation of cyclooligoarsines, supporting the formulation of the bonding in 4 as base-stabilized arsinidene R3P→AsR.
Co-reporter:Matthew J. Ray, Michael Bühl, Laurence J. Taylor, Kasun S. Athukorala Arachchige, Alexandra M. Z. Slawin, and Petr Kilian
Inorganic Chemistry 2014 Volume 53(Issue 16) pp:8538-8547
Publication Date(Web):July 29, 2014
DOI:10.1021/ic501142v
Coordination chemistry of an acenaphthene peri-backbone-supported phosphino-phosphonium chloride (1) was investigated, revealing three distinct modes of reactivity. The reaction of 1 with Mo(CO)4(nor) gives the Mo(0) complex [(1)Mo(CO)4Cl] (2), in which the ligand 1 exhibits monodentate coordination through the phosphine donor and the P–P bond is retained. PtCl2(cod) reacts with the chloride and triflate salts of 1 to form a mononuclear complex [(1Cl)PtCl2] (3) and a binuclear complex [((1Cl)PtCl)2][2TfO] (4), respectively. In both of these complexes, the platinum center adds across the P–P bond, and subsequent chloride transfer to the phosphenium center results in phosphine-chlorophosphine bidentate coordination. [((1)PdCl)2] (5) was isolated from the reaction of 1 and Pd2(dba)3 (dba = dibenzylideneacetone). Oxidative addition to palladium(0) results in a heteroleptic phosphine bridging phosphide coordination to the Pd(II) center. In addition, reaction of 1 with BH3·SMe2 leads to the bis(borane) adduct of the corresponding mixed tertiary/secondary phosphine (6), with BH3 acting as both a reducing agent and a Lewis acid. The new compounds were fully characterized, including X-ray diffraction. The ligand properties of 1 and related bonding issues are discussed with help of DFT computations.
Co-reporter:Brian A. Surgenor, Brian A. Chalmers, Kasun S. Athukorala Arachchige, Alexandra M. Z. Slawin, J. Derek Woollins, Michael Bühl, and Petr Kilian
Inorganic Chemistry 2014 Volume 53(Issue 13) pp:6856-6866
Publication Date(Web):June 9, 2014
DOI:10.1021/ic500697m
The reactions of peri-substitution-stabilized phosphanylidene-phosphorane 1 with [AuCl(tht)] or [PtCl2(cod)] afford binuclear complexes [((1)(AuCl)2)2] 2 and [((1)(PtCl2))2] 3, in which four electrons of the ligand are used in bonding to two metal atoms in the bridging arrangement. Reactions of 1 with [Mo(CO)4(nbd)] or (RhCl2Cp*)2 afford mononuclear complexes [(1)2Mo(CO)4] 4 and [(1)RhCl2Cp*] 5, in which two electrons of the ligand are used to form terminal complexes. Formation of these complexes disrupts the negative hyperconjugation at the P–P bond to various extents, which is mirrored by variations in their P–P bond distances (2.179(4)–2.246(4) Å). The P–P bond is ruptured upon formation of Pd diphosphene complex 6, which is likely to proceed through a phosphinidene intermediate. In air, 1 is fully oxidized to phosphonic acid 7. Reactions of 1 with chalcogens under mild conditions generally afford mixtures of products, from which the trithionated 8, dithionated 9, diselenated 10, and monotellurated 11 species were isolated. The bonding in the chalcogeno derivatives is discussed using DFT (B3LYP) and natural bond orbital analysis, which indicate a contribution from dative bonding in 8–10. The buttressing effect of the peri backbone is shown to be an essential factor in the formation of the single push–double-pull bis(borane) 13. This is demonstrated experimentally through a synthesis parallel to that used to make 13, but lacking the backbone, which leads to different products. The P–P bond distances in the reported products, as well as additional species, are correlated with Wiberg bond indices, showing very good agreement for a variety of bonding modes, including the negative hyperconjugation.
Co-reporter:Matthew J. Ray, Rebecca A. M. Randall, Kasun S. Athukorala Arachchige, Alexandra M. Z. Slawin, Michael Bühl, Tomas Lebl, and Petr Kilian
Inorganic Chemistry 2013 Volume 52(Issue 8) pp:4346-4359
Publication Date(Web):March 27, 2013
DOI:10.1021/ic3024875
Coupling of two acenaphthene backbones through a phosphorus atom in a geminal fashion gives the first geminally bis(peri-substituted) tridentate phosphine 1. The rigid nature of the aromatic backbone and overall crowding of the molecule result in a rather inflexible ligand, with the three phosphorus atoms forming a relatively compact triangular cluster. Phosphine 1 displays restricted dynamics on an NMR time scale, which leads to the anisochronicity of all three phosphorus nuclei at low temperatures. Strained bis- and tris(sulfides) 2 and 3 and the bis(selenide) 4 have been isolated from the reaction of 1 with sulfur and selenium, respectively. These chalcogeno derivatives display pronounced in-plane and out-of-plane distortions of the aromatic backbones, indicating the limits of their angular distortions. In addition, we report metal complexes with tetrahedral [(1)Cu(MeCN)][BF4] (5), square planar [(1)PtCl][Cl] (6), trigonal bipyramidal [(1)FeCl2] (7), and octahedral fac-[(1)Mo(CO)3] (8) geometries. In all of these complexes the tris(phosphine) backbone is distorted, however to a significantly smaller extent than that in the mentioned chalcogenides 2–4. Complexes 5 and 8 show fluxionality in 31P and 1H NMR. All new compounds 1–8 were fully characterized, and their crystal structures are reported. Conclusions from dynamic NMR observations were augmented by DFT calculations.
Co-reporter:Conor G. E. Fleming, Alexandra M. Z. Slawin, Kasun S. Athukorala Arachchige, Rebecca Randall, Michael Bühl and Petr Kilian
Dalton Transactions 2013 vol. 42(Issue 5) pp:1437-1450
Publication Date(Web):06 Nov 2012
DOI:10.1039/C2DT31971C
Reaction chemistry of an extremely sterically encumbered phosphinic chloride (Mes*)2P(O)Cl (Mes* = 2,4,6-tri-t-butylphenyl, supermesityl) was investigated. This compound, as well as other compounds bearing two supermesityl groups placed geminally at the central phosphorus atom, shows extremely low reactivity at the phosphorus centre. Nevertheless, some synthetically significant transformations were possible. Reduction with hydridic reagents under forcing conditions yielded the phosphine oxide (Mes*)2P(O)H and a secondary phosphine Mes*(2,4-tBu2C6H3)PH. Deprotonation of (Mes*)2P(O)H gave the corresponding phosphinite, which afforded very crowded tertiary phosphine oxides (Mes*)2P(O)R (R = Me and Et) on reactions with electrophiles. While the reaction of the phosphine Mes*(2,4-tBu2C6H3)PH with sulfur was surprisingly facile (although under forcing conditions), we have been unable to chlorinate or deprotonate this phosphine. All new compounds were fully characterised with multinuclear NMR, IR, Raman, MS, microanalyses and single crystal X-ray diffraction. Our computations (B3LYP and M06-2X level) show that strain energies of (synthetically accessible) geminally substituted compounds are extremely high (180 to 250 kJ mol−1), the majority of the strain is stored as boat distortions to the phenyl rings in Mes* substituents.
Co-reporter:Matthew J. Ray, Alexandra M. Z. Slawin, Michael Bühl, and Petr Kilian
Organometallics 2013 Volume 32(Issue 12) pp:3481-3492
Publication Date(Web):June 5, 2013
DOI:10.1021/om400259h
The clean reaction of 5-lithio-6-(diisopropylphosphino)acenaphthene with dichlorophosphines RPCl2 gives the peri-substituted phosphino-phosphonium salts [Acenap(PiPr2)(PR)]+Cl– (2, R = Ph; 3, R = Fc; 4, R = NMe2; 5, R = iPr; Acenap = acenaphthene-5,6-diyl). Their ionic structure is maintained in solution and in the solid state. The reduction of 2 and 3 with LiAlH4 led to the formation of mixed tertiary/secondary chelating bis(phosphines) Acenap(PiPr2)(PRH) (6 and 7), which were subsequently reacted with PtCl2(cod) to give the complexes [(6)/(7)PtCl2] (8 and 9). Reaction of 2 and 3 with a large excess of MeOTf at elevated temperature gave the chiral 1,2-diphosphoniums [Acenap(PiPr2)(PRMe)]2+([TfO]−)2 (10 and 11), which were reduced with LiAlH4 to the heteroleptic bis(phosphines) Acenap(PiPr2)(PRMe) (12 and 13); these were then reacted with [(nor)Mo(CO)4] to give the complexes [(12)/(13)Mo(CO)4] (14 and 15). The heteroleptic bis(phosphines) 6, 7, 12, and 13 display large through-space couplings (formally 4JPP = 163–199 Hz), comparable in magnitude to 1JPP couplings observed in phosphino-phosphonium salts 2–5 (303–412 Hz). Single-crystal X-ray structures of 2, 3, 7–9, 14, and 15 are reported.
Co-reporter:Brian A. Surgenor; Michael Bühl; Alexra M. Z. Slawin; J. Derek Woollins ;Dr. Petr Kilian
Angewandte Chemie International Edition 2012 Volume 51( Issue 40) pp:10150-10153
Publication Date(Web):
DOI:10.1002/anie.201204998
Co-reporter:Brian A. Surgenor; Michael Bühl; Alexra M. Z. Slawin; J. Derek Woollins ;Dr. Petr Kilian
Angewandte Chemie 2012 Volume 124( Issue 40) pp:10297-10300
Publication Date(Web):
DOI:10.1002/ange.201204998
Co-reporter:Petr Kilian, Fergus R. Knight, J. Derek Woollins
Coordination Chemistry Reviews 2011 Volume 255(11–12) pp:1387-1413
Publication Date(Web):June 2011
DOI:10.1016/j.ccr.2011.01.015
The synthetic aspects of chemistry of ligands based on naphthalene peri-substituted by heavier Group 15 elements (P, As, Sb, Bi) or Group 16 elements (S, Se, Te) are discussed in this review. An overview of coordination chemistry of these ligands is also given. In general, the area is dominated by bis(phosphines) Nap(PR2)2 and dithiolates Nap(SR)2 (Nap = naphthalene-1,8-diyl), and most of the ligands act with chelating rigid C3-backbones. Whilst all known bis(phosphine) complexes with Ni, Pd and Pt contain unmodified Nap(PR2)2 moieties, the reactions with a variety of metal carbonyls sometimes result in P–C bond cleavage within the ligand. A range of gold complexes with Nap(PR2)2 ligands have been investigated for material applications. NapP2 ligands other than phosphines are also described, these include 1,2-diphosphaacenaphthenes, bis(phosphonites) and bis(phosphine oxides). Group 16 peri-dichalcogenolates used as ligands include NapS2, NapSe2 and NapSSe systems, but no tellurium congeners. Heterodentate ligands discussed in this review include those with NapPN, NapPO, NapPS, NapPF, NapPC and NapSN motifs. Ligands with heavier Group 15 donor atoms (NapAs2, NapSb2) are also reported. All possible oxides of the dithioles (monooxide to tetraoxide) as ligands are also discussed. Areas of interest for further work are outlined.The coordination chemistry of ligands based on naphthalene peri-substituted by heavier Group 15 (P, As, Sb, Bi) or Group 16 (O, S, Se, Te) elements, is dominated by chelating bis(phosphines) Nap(PR2)2 and dithiolates (NapS2)2 (Nap = naphthalene-1,8-diyl).
Co-reporter:Kenneth M. Armstrong
European Journal of Inorganic Chemistry 2011 Volume 2011( Issue 13) pp:2138-2147
Publication Date(Web):
DOI:10.1002/ejic.201100046
Abstract
Triaryl phosphates were synthesized from white phosphorus and phenols under aerobic conditions and in the presence of iron catalysts and iodine. Full conversion to phosphates was achieved without the use of chlorine, and the reactions do not produce acid waste. Triphenyl phosphate, tritolyl phosphate and tris(2,4-di-tert-butyl)phenyl phosphate were synthesized by this method with high selectivities. Various iron(III) diketonates were used to catalyze the conversion. Mechanistic studies showed that the reaction proceeds by formation of PI3, then O=PI(OPh)2 before the final formation of the phosphate. The nucleophilic substitution of O=PI(OPh)2 with phenol to form O=P(OPh)3 was found to be the rate-limiting step.
Co-reporter:Dr. Petr Kilian;Dr. Fergus R. Knight ;Dr. J. Derek Woollins
Chemistry - A European Journal 2011 Volume 17( Issue 8) pp:2302-2328
Publication Date(Web):
DOI:10.1002/chem.201001750
Abstract
Synthetic and bonding aspects of heavier Group 15 (P, As, Sb, Bi) and 16 (S, Se, Te) peri-substituted naphthalenes, are discussed in this review. An important and unifying feature of the chemistry of these systems is the lively discussion about the nature of the interaction between peri-atoms. Are atoms bonded when they are closer than the sum of their van der Waals radii? Is there any (weak) bonding, or just a strained repulsive interaction? Positioning atoms of Group 15 and 16 at the naphthalene 1,8-positions provides leading systems with which to study these bonding issues.
Co-reporter:D. M. Upulani K. Somisara;Dr. Michael Bühl;Dr. Tomas Lebl;Dr. Neville V. Richardson;Dr. Alexra M. Z. Slawin;Dr. J. Derek Woollins ;Dr. Petr Kilian
Chemistry - A European Journal 2011 Volume 17( Issue 9) pp:2666-2677
Publication Date(Web):
DOI:10.1002/chem.201002259
Abstract
Syntheses and full characterisation data (including single crystal diffraction) of three 1,2-diphosphonium dicationic species with the naphthalene-1,8-diyl (Nap) backbone are reported. The oxidation of Nap[P(NMe2)2]2 with P2I4 to its 1,2-dication was achieved. meso- and rac-forms of “all carbon” 1,2-diphosphonium dications were obtained in good yields and purity by double alkylation of the parent diphosphine (1,2-diphenyl-1,2-diphosphaacenaphthene) with methyl triflate or trimethyloxonium tetrafluoroborate. Each methylating reagent produces one of the rac- or meso-forms of the dication diastereospecifically. Structural parameters of the new dications are discussed with respect to other phosphorus 1,2-dications. DFT (B3LYP) computations revealed the significant role of the naphthalene backbone in stabilisation of the dicationic motif and helped to assess the energy cost of the steric clash of a variety of groups attached to the peri-positions of naphthalene. The synthesis and single crystal X-ray data of the extremely crowded Nap[P(Se)(OiPr)2]2 are discussed, and are contrasted with the unsuccessful synthesis of Nap(PtBu2)2 from NapLi2 and ClPtBu2.
Co-reporter:Piotr Wawrzyniak, Alexandra M. Z. Slawin, J. Derek Woollins and Petr Kilian
Dalton Transactions 2010 vol. 39(Issue 1) pp:85-92
Publication Date(Web):05 Nov 2009
DOI:10.1039/B916425A
Syntheses of heteroleptic 1,8-bis(phosphino)naphthalenes and 5,6-bis(phosphino)acenaphthenes were attempted using several synthetic strategies. Reaction of aryllithium with triphenylphosphite gave ArP(OPh)2 (Ar = substituted naphthalene or acenaphthene), which was transformed into ArP(CF3)2, using a nucleophilic trifluoromethylation reaction with Me3SiCF3/CsF. The importance of the correct choice of solvent for the trifluoromethylation reactions is discussed. The incompatibility of ArP(CF3)2 with organolithium hampered the attachment of the second phosphine functionality to the organic backbone. Tetraphenoxyethylene was obtained in a small amount as a side product in the trifluoromethylation reaction. Selected new compounds were characterized by single-crystal X-ray diffraction.
Co-reporter:Piotr Wawrzyniak, Amy L. Fuller, Alexandra M. Z. Slawin and Petr Kilian
Inorganic Chemistry 2009 Volume 48(Issue 6) pp:2500-2506
Publication Date(Web):February 13, 2009
DOI:10.1021/ic801833a
The reaction of 5-diphenoxyphosphanyl-6-diisopropylphosphinoacenaphthene 12 with chlorotrimethylsilane unexpectedly gave a phosphonium-phosphine compound 13, containing the structural motif of four phosphorus atoms connected in a chain. To explain the mechanism of this complex transformation, a proposed intermediate 5-dichlorophosphino-6-diisopropylphosphinoacenaphthene 14 was synthesized by an alternative method. The two (formally) phosphine environments in 14 form an intramolecular donor−acceptor (phosphonium-phosphoranide) complex, stable at room temperature in the solid state and as a solution in certain solvents. A 31P NMR mechanistic study showed that, despite the presence of a rigid acenaphthene backbone, 14 is unstable in the presence of nucleophiles and disproportionates into 13 and other phosphorus containing products. Both 13 and 14 have been crystallographically characterized.
Co-reporter:Piotr Wawrzyniak, Alexandra M. Z. Slawin, Amy L. Fuller, J. Derek Woollins and Petr Kilian
Dalton Transactions 2009 (Issue 38) pp:7883-7884
Publication Date(Web):21 Aug 2009
DOI:10.1039/B916166J
Using a novel strategy, tertiary phosphines based on single atom peri-bridged acenaphthenes were prepared and characterized by multinuclear NMR and by X-ray diffraction.
Co-reporter:Petr Kilian and Alexandra M. Z. Slawin
Dalton Transactions 2007 (Issue 30) pp:3289-3296
Publication Date(Web):15 Jun 2007
DOI:10.1039/B705286C
A series of 1,8,9-tris(phosphorus) substituted anthracenes was synthesised and fully characterised. Their molecular geometry is such that the middle phosphorus atom is in a highly congested space, with terminal phosphorus atoms in close proximity. The middle phosphorus atom accepts electron density from none, one or both terminal phosphorus atoms to form P–P bonds. Thus phosphino (repulsively interacting), metaphosphonato (single donor stabilised) as well as phosphenium (doubly phosphine donor stabilised) environments around the phosphorus atom in position 9 were synthesized and structurally characterised. Observed P⋯P distances range from 2.22 to 3.27 Å.
Co-reporter:Petr Kilian and Alexandra M. Z. Slawin
Dalton Transactions 2007(Issue 30) pp:
Publication Date(Web):
DOI:10.1039/B705286C
Co-reporter:Piotr Wawrzyniak, Alexandra M. Z. Slawin, Amy L. Fuller, J. Derek Woollins and Petr Kilian
Dalton Transactions 2009(Issue 38) pp:NaN7884-7884
Publication Date(Web):2009/08/21
DOI:10.1039/B916166J
Using a novel strategy, tertiary phosphines based on single atom peri-bridged acenaphthenes were prepared and characterized by multinuclear NMR and by X-ray diffraction.
Co-reporter:Laurence J. Taylor, Brian A. Surgenor, Piotr Wawrzyniak, Matthew J. Ray, David B. Cordes, Alexandra M. Z. Slawin and Petr Kilian
Dalton Transactions 2016 - vol. 45(Issue 5) pp:NaN1986-1986
Publication Date(Web):2015/08/21
DOI:10.1039/C5DT02539G
Bis(borane) adducts Acenap(PiPr2·BH3)(PRH·BH3) (Acenap = acenaphthene-5,6-diyl; 4a, R = Ph; 4b, R = ferrocenyl, Fc; 4c, R = H) were synthesised by the reaction of excess H3B·SMe2 with either phosphino-phosphonium salts [Acenap(PiPr2)(PR)]+Cl− (1a, R = Ph; 1b, R = Fc), or bis(phosphine) Acenap(PiPr2)(PH2) (3). Bis(borane) adducts 4a–c were found to undergo dihydrogen elimination at room temperature, this spontaneous catalyst-free phosphine-borane dehydrocoupling yields BH2 bridged species Acenap(PiPr2)(μ-BH2)(PR·BH3) (5a, R = Ph; 5b, R = Fc; 5c, R = H). Thermolysis of 5c results in loss of the terminal borane moiety to afford Acenap(PiPr2)(μ-BH2)(PH) (14). Single crystal X-ray structures of 3, 4b and 5a–c are reported.
Co-reporter:Conor G. E. Fleming, Alexandra M. Z. Slawin, Kasun S. Athukorala Arachchige, Rebecca Randall, Michael Bühl and Petr Kilian
Dalton Transactions 2013 - vol. 42(Issue 5) pp:NaN1450-1450
Publication Date(Web):2012/11/06
DOI:10.1039/C2DT31971C
Reaction chemistry of an extremely sterically encumbered phosphinic chloride (Mes*)2P(O)Cl (Mes* = 2,4,6-tri-t-butylphenyl, supermesityl) was investigated. This compound, as well as other compounds bearing two supermesityl groups placed geminally at the central phosphorus atom, shows extremely low reactivity at the phosphorus centre. Nevertheless, some synthetically significant transformations were possible. Reduction with hydridic reagents under forcing conditions yielded the phosphine oxide (Mes*)2P(O)H and a secondary phosphine Mes*(2,4-tBu2C6H3)PH. Deprotonation of (Mes*)2P(O)H gave the corresponding phosphinite, which afforded very crowded tertiary phosphine oxides (Mes*)2P(O)R (R = Me and Et) on reactions with electrophiles. While the reaction of the phosphine Mes*(2,4-tBu2C6H3)PH with sulfur was surprisingly facile (although under forcing conditions), we have been unable to chlorinate or deprotonate this phosphine. All new compounds were fully characterised with multinuclear NMR, IR, Raman, MS, microanalyses and single crystal X-ray diffraction. Our computations (B3LYP and M06-2X level) show that strain energies of (synthetically accessible) geminally substituted compounds are extremely high (180 to 250 kJ mol−1), the majority of the strain is stored as boat distortions to the phenyl rings in Mes* substituents.
Co-reporter:Piotr Wawrzyniak, Alexandra M. Z. Slawin, J. Derek Woollins and Petr Kilian
Dalton Transactions 2010 - vol. 39(Issue 1) pp:NaN92-92
Publication Date(Web):2009/11/05
DOI:10.1039/B916425A
Syntheses of heteroleptic 1,8-bis(phosphino)naphthalenes and 5,6-bis(phosphino)acenaphthenes were attempted using several synthetic strategies. Reaction of aryllithium with triphenylphosphite gave ArP(OPh)2 (Ar = substituted naphthalene or acenaphthene), which was transformed into ArP(CF3)2, using a nucleophilic trifluoromethylation reaction with Me3SiCF3/CsF. The importance of the correct choice of solvent for the trifluoromethylation reactions is discussed. The incompatibility of ArP(CF3)2 with organolithium hampered the attachment of the second phosphine functionality to the organic backbone. Tetraphenoxyethylene was obtained in a small amount as a side product in the trifluoromethylation reaction. Selected new compounds were characterized by single-crystal X-ray diffraction.

![Phosphonic dichloride, [8-(tetrachlorophosphoranyl)-1-naphthalenyl]-](http://img.cochemist.com/ccimg/635000/634929-30-9.png)
![Phosphonic dichloride, [8-(tetrachlorophosphoranyl)-1-naphthalenyl]-](http://img.cochemist.com/ccimg/635000/634929-30-9_b.png)
![[1,1'-Biphenyl]-2,2'-dithiol](http://img.cochemist.com/ccimg/43300/43216-01-9.png)
![[1,1'-Biphenyl]-2,2'-dithiol](http://img.cochemist.com/ccimg/43300/43216-01-9_b.png)
![Naphtho[1,8-cd]-1,2-dithiole, 1,1,2-trioxide](http://img.cochemist.com/ccimg/57900/57821-65-5.png)
![Naphtho[1,8-cd]-1,2-dithiole, 1,1,2-trioxide](http://img.cochemist.com/ccimg/57900/57821-65-5_b.png)
![Phosphonic dichloride, [8-(dichlorophosphino)-1-naphthalenyl]-](http://img.cochemist.com/ccimg/522000/521959-79-5.png)
![Phosphonic dichloride, [8-(dichlorophosphino)-1-naphthalenyl]-](http://img.cochemist.com/ccimg/522000/521959-79-5_b.png)
![Dibenzo[c,e][1,2]diselenin](http://img.cochemist.com/ccimg/91600/91538-47-5.png)
![Dibenzo[c,e][1,2]diselenin](http://img.cochemist.com/ccimg/91600/91538-47-5_b.png)
![1H,3H-Naphth[1,8-cd][1,2,6]oxadiphosphorin, 1-hydroxy-, 1,3-dioxide](http://img.cochemist.com/ccimg/683900/683809-59-8.png)
![1H,3H-Naphth[1,8-cd][1,2,6]oxadiphosphorin, 1-hydroxy-, 1,3-dioxide](http://img.cochemist.com/ccimg/683900/683809-59-8_b.png)
![Phosphonic acid, mono[2,4-bis(1,1-dimethylethyl)phenyl] ester](http://img.cochemist.com/ccimg/738600/738537-08-1.png)
![Phosphonic acid, mono[2,4-bis(1,1-dimethylethyl)phenyl] ester](http://img.cochemist.com/ccimg/738600/738537-08-1_b.png)
![DIBENZO[C,E][1,2]DITHIIN, 5,5,6-TRIOXIDE](http://img.cochemist.com/ccimg/63100/63059-28-9.png)
![DIBENZO[C,E][1,2]DITHIIN, 5,5,6-TRIOXIDE](http://img.cochemist.com/ccimg/63100/63059-28-9_b.png)
![DIBENZO[C,E][1,2]DITHIIN, 5-OXIDE, (-)-](http://img.cochemist.com/ccimg/49900/49833-13-8.png)
![DIBENZO[C,E][1,2]DITHIIN, 5-OXIDE, (-)-](http://img.cochemist.com/ccimg/49900/49833-13-8_b.png)


![Naphtho[1,8-cd]-1,2-dithiole, 3,8-bis(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/350300/350253-68-8.png)
![Naphtho[1,8-cd]-1,2-dithiole, 3,8-bis(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/350300/350253-68-8_b.png)
![Dinaphtho[1,2-c:2',1'-e][1,2]dithiin](http://img.cochemist.com/ccimg/300/213-52-5.png)
![Dinaphtho[1,2-c:2',1'-e][1,2]dithiin](http://img.cochemist.com/ccimg/300/213-52-5_b.png)
![NAPHTHO[1,8-CD]-1,2-DITHIOLE, 1,1-DIOXIDE](http://img.cochemist.com/ccimg/40300/40227-43-8.png)
![NAPHTHO[1,8-CD]-1,2-DITHIOLE, 1,1-DIOXIDE](http://img.cochemist.com/ccimg/40300/40227-43-8_b.png)
![1,3-EPITHIO-1H,3H-NAPHTHO[1,8-CD][1,2,6]THIADIPHOSPHORIN, 1,3-DISULFIDE](http://img.cochemist.com/ccimg/115600/115505-46-9.png)
![1,3-EPITHIO-1H,3H-NAPHTHO[1,8-CD][1,2,6]THIADIPHOSPHORIN, 1,3-DISULFIDE](http://img.cochemist.com/ccimg/115600/115505-46-9_b.png)
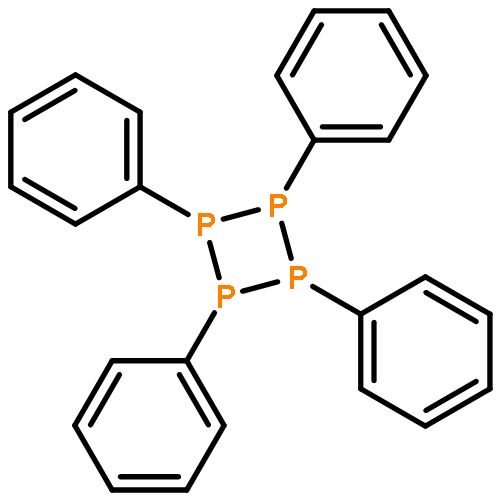
![POLY(1,2-DIHYDRONAPHTHO[1,8-CD]-1,2-DIPHOSPHOLE-1,2-DIYL)](http://img.cochemist.com/ccimg/522000/521959-80-8.png)
![POLY(1,2-DIHYDRONAPHTHO[1,8-CD]-1,2-DIPHOSPHOLE-1,2-DIYL)](http://img.cochemist.com/ccimg/522000/521959-80-8_b.png)
![PHOSPHONOUS DICHLORIDE, [2,4,6-TRIS(1,1-DIMETHYLETHYL)PHENYL]-](http://img.cochemist.com/ccimg/79100/79074-00-3.png)
![PHOSPHONOUS DICHLORIDE, [2,4,6-TRIS(1,1-DIMETHYLETHYL)PHENYL]-](http://img.cochemist.com/ccimg/79100/79074-00-3_b.png)
![dibenzo[c,e][1,2]dithiine](http://img.cochemist.com/ccimg/300/230-26-2.png)
![dibenzo[c,e][1,2]dithiine](http://img.cochemist.com/ccimg/300/230-26-2_b.png)



![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)


![Phosphinic chloride, bis[2,4,6-tris(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/73600/73557-52-5.png)
![Phosphinic chloride, bis[2,4,6-tris(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/73600/73557-52-5_b.png)









![Phosphine oxide, [2,4,6-tris(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/86600/86539-32-4.png)
![Phosphine oxide, [2,4,6-tris(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/86600/86539-32-4_b.png)
![Phosphine, methyl[2,4,6-tris(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/86600/86539-33-5.png)
![Phosphine, methyl[2,4,6-tris(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/86600/86539-33-5_b.png)







![Naphtho[1,8-cd]-1,2-dithiole,1-oxide](http://img.cochemist.com/ccimg/49900/49833-12-7.png)
![Naphtho[1,8-cd]-1,2-dithiole,1-oxide](http://img.cochemist.com/ccimg/49900/49833-12-7_b.png)


![Phosphine oxide, bis[2,4,6-tris(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/105600/105563-33-5.png)
![Phosphine oxide, bis[2,4,6-tris(1,1-dimethylethyl)phenyl]-](http://img.cochemist.com/ccimg/105600/105563-33-5_b.png)





![Naphtho[1,8-cd]-1,2-dithiole](http://img.cochemist.com/ccimg/300/209-22-3.png)
![Naphtho[1,8-cd]-1,2-dithiole](http://img.cochemist.com/ccimg/300/209-22-3_b.png)