Co-reporter:Vassilios Bavetsias; Rachel M. Lanigan; Gian Filippo Ruda; Butrus Atrash; Mark G. McLaughlin; Anthony Tumber; N. Yi Mok; Yann-Vaï Le Bihan; Sally Dempster; Katherine J. Boxall; Fiona Jeganathan; Stephanie B. Hatch; Pavel Savitsky; Srikannathasan Velupillai; Tobias Krojer; Katherine S. England; Jimmy Sejberg; Ching Thai; Adam Donovan; Akos Pal; Giuseppe Scozzafava; James M. Bennett; Akane Kawamura; Catrine Johansson; Aleksandra Szykowska; Carina Gileadi; Nicola A. Burgess-Brown; Frank von Delft; Udo Oppermann; Zoe Walters; Janet Shipley; Florence I. Raynaud; Susan M. Westaway◆; Rab K. Prinjha◆; Oleg Fedorov; Rosemary Burke; Christopher J. Schofield; Isaac M. Westwood; Chas Bountra; Susanne Müller; Rob L. M. van Montfort; Paul E. Brennan;Julian Blagg
Journal of Medicinal Chemistry 2016 Volume 59(Issue 4) pp:1388-1409
Publication Date(Web):January 7, 2016
DOI:10.1021/acs.jmedchem.5b01635
We report the discovery of N-substituted 4-(pyridin-2-yl)thiazole-2-amine derivatives and their subsequent optimization, guided by structure-based design, to give 8-(1H-pyrazol-3-yl)pyrido[3,4-d]pyrimidin-4(3H)-ones, a series of potent JmjC histone N-methyl lysine demethylase (KDM) inhibitors which bind to Fe(II) in the active site. Substitution from C4 of the pyrazole moiety allows access to the histone peptide substrate binding site; incorporation of a conformationally constrained 4-phenylpiperidine linker gives derivatives such as 54j and 54k which demonstrate equipotent activity versus the KDM4 (JMJD2) and KDM5 (JARID1) subfamily demethylases, selectivity over representative exemplars of the KDM2, KDM3, and KDM6 subfamilies, cellular permeability in the Caco-2 assay, and, for 54k, inhibition of H3K9Me3 and H3K4Me3 demethylation in a cell-based assay.
Co-reporter:Vassilios Bavetsias, Yolanda Pérez-Fuertes, Patrick J. McIntyre, Butrus Atrash, Magda Kosmopoulou, Lisa O’Fee, Rosemary Burke, Chongbo Sun, Amir Faisal, Katherine Bush, Sian Avery, Alan Henley, Florence I. Raynaud, Spiros Linardopoulos, Richard Bayliss, Julian Blagg
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 19) pp:4203-4209
Publication Date(Web):1 October 2015
DOI:10.1016/j.bmcl.2015.08.003
Introduction of a 1-benzyl-1H-pyrazol-4-yl moiety at C7 of the imidazo[4,5-b]pyridine scaffold provided 7a which inhibited a range of kinases including Aurora-A. Modification of the benzyl group in 7a, and subsequent co-crystallisation of the resulting analogues with Aurora-A indicated distinct differences in binding mode dependent upon the pyrazole N-substituent. Compounds 7a and 14d interact with the P-loop whereas 14a and 14b engage with Thr217 in the post-hinge region. These crystallographic insights provide options for the design of compounds interacting with the DFG motif or with Thr217.Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Vassilios Bavetsias ; Amir Faisal ; Simon Crumpler ; Nathan Brown ; Magda Kosmopoulou ; Amar Joshi ; Butrus Atrash ; Yolanda Pérez-Fuertes ; Jessica A. Schmitt ; Katherine J. Boxall ; Rosemary Burke ; Chongbo Sun ; Sian Avery ; Katherine Bush ; Alan Henley ; Florence I. Raynaud ; Paul Workman ; Richard Bayliss ; Spiros Linardopoulos ;Julian Blagg
Journal of Medicinal Chemistry 2013 Volume 56(Issue 22) pp:9122-9135
Publication Date(Web):November 6, 2013
DOI:10.1021/jm401115g
Aurora-A differs from Aurora-B/C at three positions in the ATP-binding pocket (L215, T217, and R220). Exploiting these differences, crystal structures of ligand–Aurora protein interactions formed the basis of a design principle for imidazo[4,5-b]pyridine-derived Aurora-A-selective inhibitors. Guided by a computational modeling approach, appropriate C7-imidazo[4,5-b]pyridine derivatization led to the discovery of highly selective inhibitors, such as compound 28c, of Aurora-A over Aurora-B. In HCT116 human colon carcinoma cells, 28c and 40f inhibited the Aurora-A L215R and R220K mutants with IC50 values similar to those seen for the Aurora-A wild type. However, the Aurora-A T217E mutant was significantly less sensitive to inhibition by 28c and 40f compared to the Aurora-A wild type, suggesting that the T217 residue plays a critical role in governing the observed isoform selectivity for Aurora-A inhibition. These compounds are useful small-molecule chemical tools to further explore the function of Aurora-A in cells.
Co-reporter:Vassilios Bavetsias ; Simon Crumpler ; Chongbo Sun ; Sian Avery ; Butrus Atrash ; Amir Faisal ; Andrew S. Moore ; Magda Kosmopoulou ; Nathan Brown ; Peter W. Sheldrake ; Katherine Bush ; Alan Henley ; Gary Box ; Melanie Valenti ; Alexis de Haven Brandon ; Florence I. Raynaud ; Paul Workman ; Suzanne A. Eccles ; Richard Bayliss ; Spiros Linardopoulos ;Julian Blagg
Journal of Medicinal Chemistry 2012 Volume 55(Issue 20) pp:8721-8734
Publication Date(Web):October 8, 2012
DOI:10.1021/jm300952s
Optimization of the imidazo[4,5-b]pyridine-based series of Aurora kinase inhibitors led to the identification of 6-chloro-7-(4-(4-chlorobenzyl)piperazin-1-yl)-2-(1,3-dimethyl-1H-pyrazol-4-yl)-3H-imidazo[4,5-b]pyridine (27e), a potent inhibitor of Aurora kinases (Aurora-A Kd = 7.5 nM, Aurora-B Kd = 48 nM), FLT3 kinase (Kd = 6.2 nM), and FLT3 mutants including FLT3-ITD (Kd = 38 nM) and FLT3(D835Y) (Kd = 14 nM). FLT3-ITD causes constitutive FLT3 kinase activation and is detected in 20–35% of adults and 15% of children with acute myeloid leukemia (AML), conferring a poor prognosis in both age groups. In an in vivo setting, 27e strongly inhibited the growth of a FLT3-ITD-positive AML human tumor xenograft (MV4–11) following oral administration, with in vivo biomarker modulation and plasma free drug exposures consistent with dual FLT3 and Aurora kinase inhibition. Compound 27e, an orally bioavailable dual FLT3 and Aurora kinase inhibitor, was selected as a preclinical development candidate for the treatment of human malignancies, in particular AML, in adults and children.
Co-reporter:Vassilios Bavetsias ; Jonathan M. Large ; Chongbo Sun ; Nathalie Bouloc ; Magda Kosmopoulou ; Mizio Matteucci ; Nicola E. Wilsher ; Vanessa Martins ; Jóhannes Reynisson ; Butrus Atrash ; Amir Faisal ; Frederique Urban ; Melanie Valenti ; Alexis de Haven Brandon ; Gary Box ; Florence I. Raynaud ; Paul Workman ; Suzanne A. Eccles ; Richard Bayliss ; Julian Blagg ; Spiros Linardopoulos ;Edward McDonald
Journal of Medicinal Chemistry 2010 Volume 53(Issue 14) pp:5213-5228
Publication Date(Web):June 21, 2010
DOI:10.1021/jm100262j
Lead optimization studies using 7 as the starting point led to a new class of imidazo[4,5-b]pyridine-based inhibitors of Aurora kinases that possessed the 1-benzylpiperazinyl motif at the 7-position, and displayed favorable in vitro properties. Cocrystallization of Aurora-A with 40c (CCT137444) provided a clear understanding into the interactions of this novel class of inhibitors with the Aurora kinases. Subsequent physicochemical property refinement by the incorporation of solubilizing groups led to the identification of 3-((4-(6-bromo-2-(4-(4-methylpiperazin-1-yl)phenyl)-3H-imidazo[4,5-b]pyridin-7-yl)piperazin-1-yl)methyl)-5-methylisoxazole (51, CCT137690) which is a potent inhibitor of Aurora kinases (Aurora-A IC50 = 0.015 ± 0.003 μM, Aurora-B IC50 = 0.025 μM, Aurora-C IC50 = 0.019 μM). Compound 51 is highly orally bioavailable, and in in vivo efficacy studies it inhibited the growth of SW620 colon carcinoma xenografts following oral administration with no observed toxicities as defined by body weight loss.
Co-reporter:Nathalie Bouloc, Jonathan M. Large, Magda Kosmopoulou, Chongbo Sun, Amir Faisal, Mizio Matteucci, Jóhannes Reynisson, Nathan Brown, Butrus Atrash, Julian Blagg, Edward McDonald, Spiros Linardopoulos, Richard Bayliss, Vassilios Bavetsias
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 20) pp:5988-5993
Publication Date(Web):15 October 2010
DOI:10.1016/j.bmcl.2010.08.091
Co-crystallisation of the imidazo[1,2-a]pyrazine derivative 15 (3-chloro-N-(4-morpholinophenyl)-6-(pyridin-3-yl)imidazo[1,2-a]pyrazin-8-amine) with Aurora-A provided an insight into the interactions of this class of compound with Aurora kinases. This led to the design and synthesis of potent Aurora-A inhibitors demonstrating up to 70-fold selectivity in cell-based Aurora kinase pharmacodynamic biomarker assays.
Co-reporter:Camilo E. Quevedo, Vassilios Bavetsias, Edward McDonald
Tetrahedron Letters 2009 50(21) pp: 2481-2483
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.03.034
Co-reporter:Elisa A. Henderson, Vassilios Bavetsias, Davinder S. Theti, Stuart C. Wilson, Rainer Clauss, Ann L. Jackman
Bioorganic & Medicinal Chemistry 2006 Volume 14(Issue 14) pp:5020-5042
Publication Date(Web):15 July 2006
DOI:10.1016/j.bmc.2006.03.001
The α-FR has been reported to be overexpressed in many carcinomas, in particular those of the ovary and uterus. The high expression of α-FR in some tumours compared with normal tissues has been exploited over the last decade for folate-mediated targeting of macromolecules, anticancer drugs, imaging agents and nucleic acids to cancer cells. CB300638, a cyclopenta[g]quinazoline-based inhibitor of thymidylate synthase (TS), has been reported to have high affinity for the receptor and selectivity for α-FR overexpressing tumour cell lines. In this study, the structural features of the molecule, in particular modifications at the 2-position, have been investigated with respect to TS inhibition, affinity for the α-FR and reduced folate carrier (RFC) and activity in A431-FBP cells (transfected with human α-FR) compared with neo-transfected A431 cells. Compounds 1a,b, 2a,b and 3a,b were synthesised utilising multistep sequences. It was found that the 2-substituent does not affect the affinity for the α-FR; however, it greatly affects selectivity for A431-FBP cells, and suggests that there are factors other than TS inhibition and α-FR affinity that are important for the activity of these compounds. Compound 2b (2-CH2OH derivative) displayed the highest selectivity for the A431-FBP cells compared with A431 cells.

![N-[2-(2-pyridinyl)-6-(1,2,4,5-tetrahydro-3h-3-benzazepin-3-yl)-4- Pyrimidinyl]-β-alanine](http://img.cochemist.com/ccimg/1373500/1373422-53-7.png)
![N-[2-(2-pyridinyl)-6-(1,2,4,5-tetrahydro-3h-3-benzazepin-3-yl)-4- Pyrimidinyl]-β-alanine](http://img.cochemist.com/ccimg/1373500/1373422-53-7_b.png)
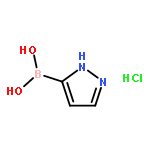
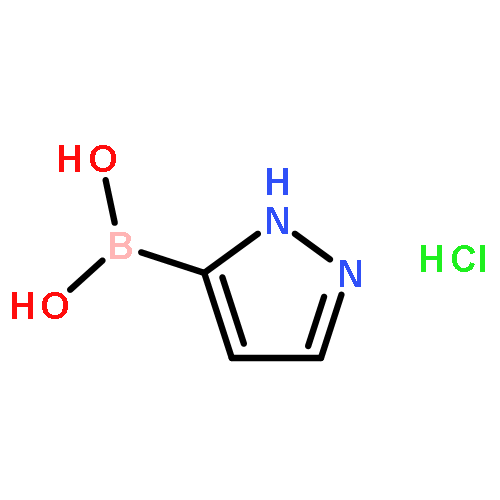
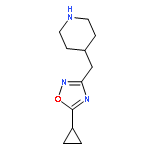
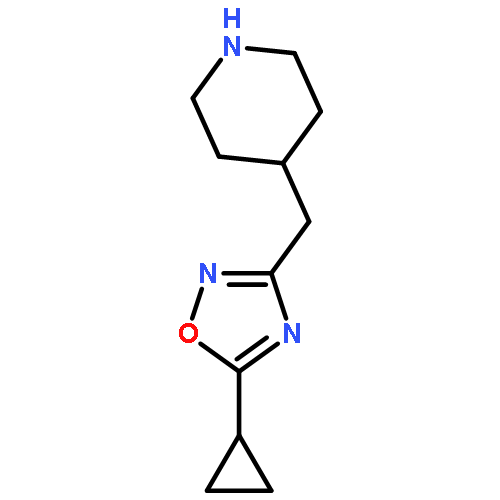


![Piperidine, 4-[(3,4-dichlorophenyl)methyl]-](http://img.cochemist.com/ccimg/220800/220772-32-7.png)
![Piperidine, 4-[(3,4-dichlorophenyl)methyl]-](http://img.cochemist.com/ccimg/220800/220772-32-7_b.png)


![Piperidine, 4-[[4-(trifluoromethyl)phenyl]methyl]-](http://img.cochemist.com/ccimg/193000/192990-03-7.png)
![Piperidine, 4-[[4-(trifluoromethyl)phenyl]methyl]-](http://img.cochemist.com/ccimg/193000/192990-03-7_b.png)
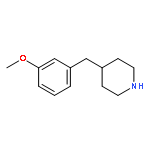
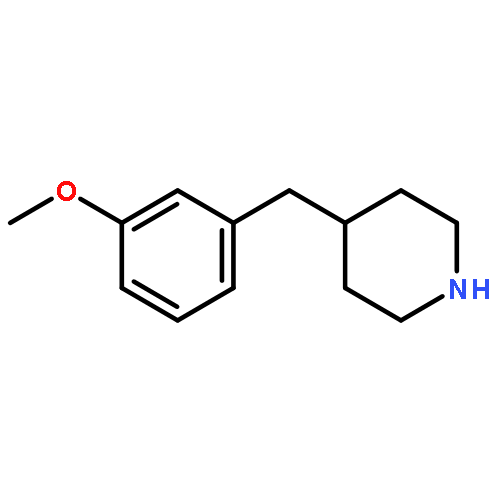
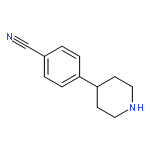
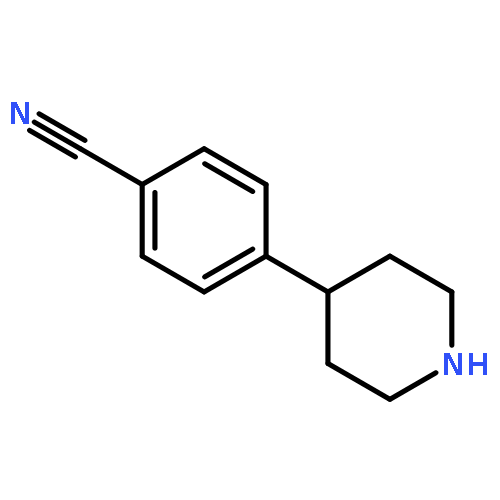
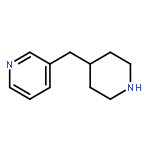
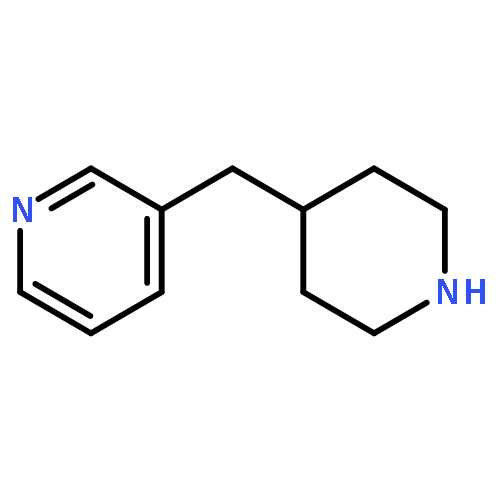
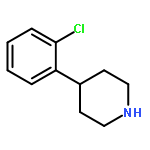

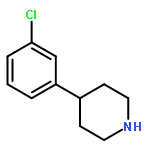
![PYRIDO[3,4-D]PYRIMIDIN-4(1H)-ONE, 8-CHLORO-](http://img.cochemist.com/ccimg/84400/84341-13-9.png)
![PYRIDO[3,4-D]PYRIMIDIN-4(1H)-ONE, 8-CHLORO-](http://img.cochemist.com/ccimg/84400/84341-13-9_b.png)
![4-BENZO[1,3]DIOXOL-5-YLMETHYL-PIPERIDINE](http://img.cochemist.com/ccimg/76700/76672-65-6.png)
![4-BENZO[1,3]DIOXOL-5-YLMETHYL-PIPERIDINE](http://img.cochemist.com/ccimg/76700/76672-65-6_b.png)
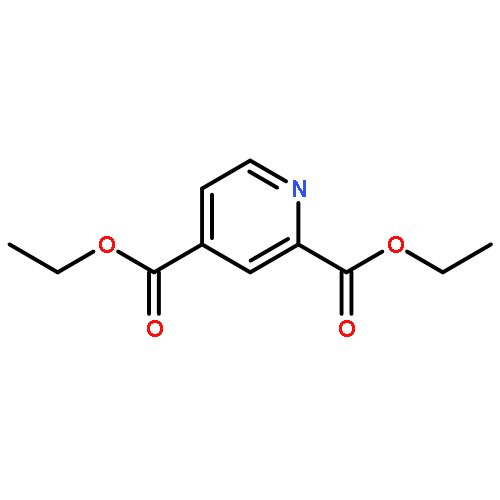
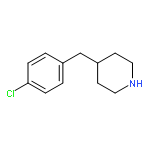
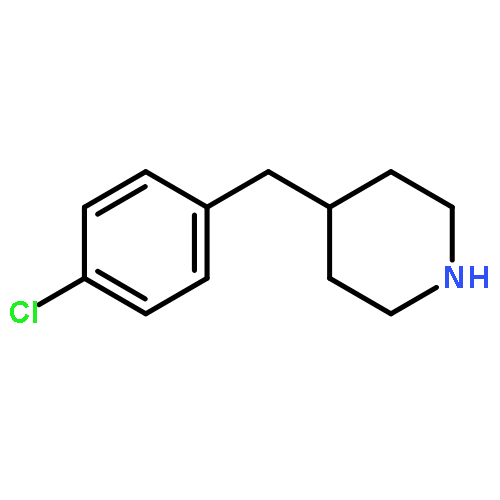
![4-[3-(TRIFLUOROMETHYL)PHENYL]PIPERIDINE](http://img.cochemist.com/ccimg/32900/32860-17-6.png)
![4-[3-(TRIFLUOROMETHYL)PHENYL]PIPERIDINE](http://img.cochemist.com/ccimg/32900/32860-17-6_b.png)