The reaction of tetrahydropyranyl (THP) ethers with triethylsilyl trifluoromethanesulfonate (TESOTf)–2,4,6-collidine proceeded via collidinium salt intermediates to give the alcohol and 4-triethylsiloxybutanal in good yields. The structure of the intermediate was confirmed by 1H NMR and FAB-MS studies and by trapping it with EtOH. The reaction was applied for mild, efficient, and highly chemoselective deprotection method of THP ethers. The characteristic feature of the reaction is that the reaction conditions are weakly basic. Then, the reaction can proceed without affecting acid-labile protecting groups. Furthermore, the intermediates from alkenol–THP ether were trapped with other alkenols to give acyclic mixed acetals, which were subjected to efficient ring-closing metathesis by using the tetrahydropyranyl unit as a tether.
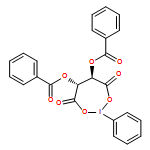
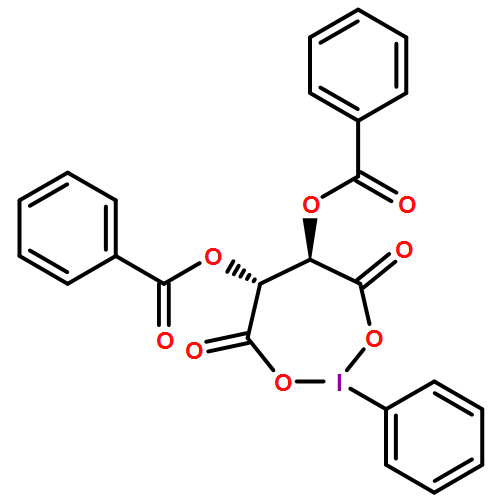
The domino reaction of 2,3-epoxy-1-alcohol derivatives, namely tetrasubstituted 2,3-epoxy-1-alcohols and 2- or 3-alkyl trisubstituted 2,3-epoxy-1-alcohols, with PhI(OCOCF3)2 in the presence of H2O is described in detail. In this reaction, several types of lactol derivatives can be directly obtained from the 2,3-epoxy-1-alcohol derivatives in a single operation. The obtained lactols were successively converted into the corresponding lactones. This reaction is applicable to the construction of optically active lactone compounds. The asymmetric total synthesis of (+)-tanikolide, an antifungal marine natural product, has been effectively achieved by using this reaction.
Doppelt oder nichts: In einer konvergenten Synthese der Titelverbindung, eines starken Inhibitors der humanen Telomerase, wurden zwei Pummerer-Arenreaktionen zum Aufbau des bisbenzannelierten Spiroketalgerüsts aus den beiden im Schema gezeigten Fragmenten genutzt. Die Methode bietet leichten Zugang zu einem Spektrum von substituierten bisbenzannelierten Spiroketalen ausgehend von Naphtholderivaten. MOM=Methoxymethyl.
Double or nothing: In a convergent synthesis of the title compound, a potent inhibitor of human telomerase, two different aromatic Pummerer-type reactions were employed to construct the pivotal bisbenzannelated spiroketal skeleton from the two fragments shown in the scheme. This methodology offers convenient access to a wide range of substituted bisbenzannelated spiroketals from naphthol derivatives. MOM=methoxymethyl.
The concise asymmetric total synthesis of scyphostatin has been achieved by condensation of the optically active cyclohexane unit, prepared from the commercially available 1,4-cyclohexadiene by our own method, and the side chain, prepared by the method developed by Hoye and Tennakoon (T. R. Hoye, M. A. Tennakoon, Org. Lett.2000, 2, 1481–1483). The modification of the epoxy cyclohexenone unit was achieved in a late stage of the total synthesis, and deprotection of the primary alcohol was conducted in the final step. During the synthesis several key reactions were attained: 1) intramolecular bromoetherification of the cyclohexadiene acetal; 2) stereoselective introduction of the tertiary alcohol, 3) deprotection of the acetal function to the aldehyde by combination with silyl triflate/2,4,6-collidine and the one-pot synthesis of the disilyl aldehyde compounds, with different types of silyl groups, from the dihydroxy acetal compounds; and 4) facile deprotection of the 2,4-dimethoxyphenylmethyl (2,4DMPM) protecting group of the primary alcohol.
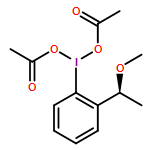
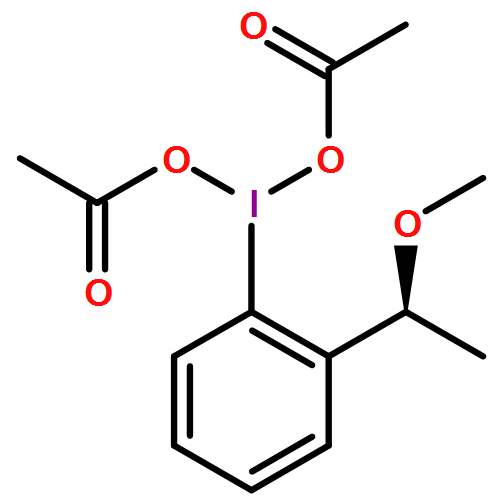
Hypervalent iodine(III) reagents are readily available, easy to handle, and have a low toxicity and similar reactivities to those of heavy metal reagents, and hence they are used for various oxidative reactions. The oxidative cleavage of alkynes or carbonyl compounds by using bis(trifluoroacetoxy)iodo(III) pentafluorobenzene (C6F5I(OCOCF3)2) has been reported.1 Herein, the efficient direct synthesis of N,O-acetal compounds as key intermediates of discorhabdin A, by the oxidative fragmentation reaction of α-amino acids or β-amino alcohols by using C6F5I(OCOCF3)2, is described.
Die Kombination macht's: Für eine neue Art dynamischer kinetischer Racematspaltung bei Allylalkoholen wird eine Kombination von Lipasen und [VO(OSiPh3)3] (1) verwendet. Die durch 1 katalysierte 1,3-Transposition der Allylalkohole lieferte eine thermodynamische Mischung zweier Regioisomere, die durch Lipasen hoch enantio- und chemoselektiv verestert wurden. Die optisch angereicherten Allylacetate wurden in hohen Ausbeuten isoliert.
In einem Schritt zu Lactolen gelangt man durch die Behandlung von 2,3-Epoxy-1-alkoholen mit Phenyliod(III)-bis(trifluoracetat) (PIFA) in Gegenwart von H2O. Somit wurde eine effiziente drei- oder vierstufige Dominosequenz entwickelt und für eine kurze Synthese von (+)-Tanikolid angewendet (siehe Schema).
Teil eines neuen und effektiven Einsatzes hypervalenter Iod(III)-Reagentien als Katalysatoren ist die Überführung von Iodarenen 1 in hypervalente Iod(III)-Verbindungen 2 mit meta-Chlorperbenzoesäure (mCPBA) als Co-Oxidans. Mit Umsatzzahlen über 70 bereitet dieses Verfahren den Weg für eine breitere Anwendung dieser nützlichen Oxidantien in der organischen Synthese.
The asymmetric total synthesis of the potent antitumor antibiotic fredericamycin A ((S)-1) was achieved by the intramolecular [4+2] cycloaddition of the silylene-protected styrene derivative (S)-7 followed by the aromatic Pummerer-type reaction of the sulfoxide (S)-5. Although we had already succeeded in the total synthesis of racemic 1 by the same approach, synthesis of its asymmetric version was more complicated than we had expected due to the difficulties involved in constructing the quaternary carbon center and the tendency of this center to undergo facile racemization. Racemization of this center during the installation of the acetylene moiety on the dione (R)-8 was the most serious aspect. Systematic studies of its DE-ring analogue (R)-25 revealed that racemization of the quaternary carbon center proceeded by a retro-aldol–aldol reaction of the initial adduct, (1R)-39 a-Li, and that the degree of racemization was dependent on the reaction temperature. The racemization process could be completely depressed by keeping the reaction temperature at −78 °C. The construction of the stereogenic quaternary carbon center was achieved by the lipase-catalyzed desymmetrization of the prochiral 1,3-diol 9 a bearing the DEF-ring moiety. These studies enabled us to attain the asymmetric total synthesis of (S)-1 while completely retaining the chiral integrity created by the enzymatic reactions.
Tetrahydrofuran units with multiple chiral centers are formed in a highly diastereoselective manner through the one-pot double intramolecular haloetherification of σ-symmetric diene acetals (see scheme). The utility of the method was demonstrated by the short asymmetric synthesis of (+)-rubrenolide.
Lactols are prepared by a single operation, in which 2,3-epoxy-1-alcohols are treated with phenyliodine(III) bis(trifluoroacetate) (PIFA) in the presence of H2O. Thus, an efficient domino three- or four-step sequence has been developed and applied to the concise synthesis of (+)-tanikolide (see scheme).
Tetrahydrofuran-Einheiten mit mehreren Chiralitätszentren entstehen hoch diastereoselektiv bei der im Schema gezeigten Eintopfreaktion aus σ-symmetrischen Dienacetalen. Die Nüzlichkeit des Verfahrens belegt die ebenfalls beschriebene kurze asymmetrische Synthese von (+)-Rubrenolid.
The combination of the water-soluble radical initiator, 2,2′-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044) and the surfactant, cetyltrimethyl-ammonium bromide (CTAB), was found to be the most suitable condition for the effective and direct synthesis of useful active thioesters (pentafluorophenyl thioesters) in water. In addition, the direct amidation of aldehydes was achieved by the addition of the amines to the thioesterification reaction mixture in water.
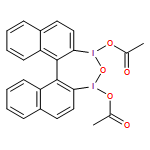
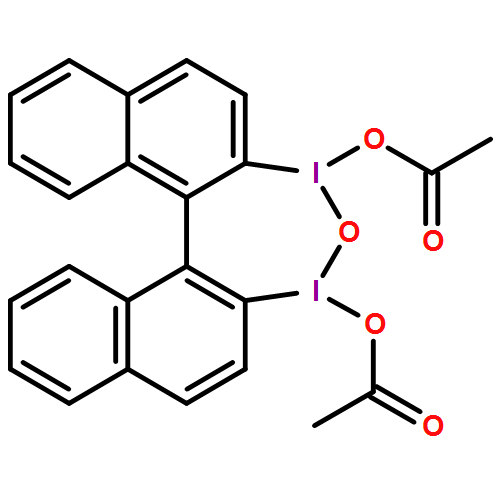
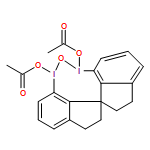
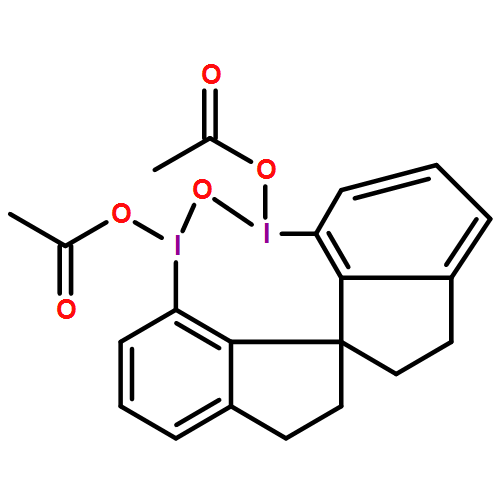
A wide range of oxidative reactions are mediated by novel, nonpolymeric, and recyclable hypervalent IIII reagents (e.g. 2). In all cases, tetraiodide 1 was recovered nearly quantitatively in pure form after a simple workup. Reoxidation of 1 to 2 with m-chloroperbenzoic acid also proceeded quantitatively, without loss of oxidative activity.
Among the various kinds of acyl donors, the 1-alkoxyvinyl esters have characteristic features, such as a high reactivity under nearly neutral conditions and the generation of neutral and volatile esters as single coproducts. Although their use in organic syntheses began in the middle of the 1950s, no significant progress has been seen. This is probably because the existing method of preparing alkoxyvinyl esters used toxic mercuric salts and was not totally applicable for those esters having functionalized acyl moieties.
We have discovered that the use of a catalytic amount of the less toxic [RuCl2(p-cymene)]2 effectively accelerates the addition of carboxylic acids to ethoxyacetylene to give ethoxyvinyl esters bearing a variety of functionalized acyl groups in high yields. This discovery has opened a new avenue for developing new reactions and new synthetic methodologies based on the design and use of these acyl donors with suitable functional groups. Such examples include (i) the installation of hydrophilic acyl moieties on biologically active compounds, (ii) asymmetric Pummerer-type reactions, (iii) aromatic Pummerer-type reactions, (iv) the lipase-catalyzed desymmetrization of symmetrical 1,3-diols, and (v) lipase-catalyzed domino reactions. Future possibilities for these acyl donors are also discussed. © 2005 The Japan Chemical Journal Forum and Wiley Periodicals, Inc. Chem Rec 4: 363–372; 2004: Published online in Wiley InterScience (www.interscience.wiley.com) DOI 10.1002/tcr.20027
A new chiral auxiliary, a 3-endo-phenyl norbornene aldehyde derivative, which is a crystalline, very stable, and easily handled, was developed for the desymmetrization of meso-1,3- and meso-1,4-diols. The key step of the method, an intramolecular bromoetherification, proceeded in a highly diastereoselective manner. A four-step sequence, 1) acetalization, 2) intramolecular bromoetherification followed by acid hydrolysis, 3) protection of the alcohol, and 4) retrobromoetherification, transformed the meso-diols into optically active derivatives. The 3-endo-phenyl norbornene aldehyde derivative was simultaneously reformed and could be used repeatedly. This is the first chemical example of a single auxiliary that is applicable for highly enantioselective desymmetrization of meso-1,3- and meso-1,4-diols; to the best of our knowledge, this is the best chemical method available for the desymmetrization of meso-1,4-diols.
The non-phenolic coupling reaction of benzyltetrahydroisoquinolines (laudanosine derivatives) by using a hypervalent iodine(III) reagent is described. In general, chemical oxidation of laudanosine gives glaucine. In contrast to general chemical oxidizing reagent systems, the novel use of reagent combination of phenyliodine bis(trifluoroacetate) (PIFA), and heteropoly acid (HPA) afforded morphinandienone alkaloids in excellent yields. In order to achieve the coupling reaction with simple reaction procedure, the use of HPA supported on silica gel instead of HPA was demonstrated and sufficient yield was exerted again. The present reagent system, PIFA/HPA, was also applied to the oxidation of other non-phenolic benzyltetrahydroisoquinolines and the high yield conversion to morphinandienones was accomplished.
Eine Vielzahl an Oxidationen gelingt mit neuartigen, nichtpolymeren und rezyklierbaren hypervalenten IIII-Reagentien (z. B. 2). In allen Fällen konnte das Tetraiodid 1 durch einfaches Aufarbeiten nahezu quantitativ in reiner Form zurückgewonnen werden. Die Reoxidation von 1 zu 2 mit m-Chlorperbenzoesäure verläuft ebenfalls quantitativ und ohne Verlust an Oxidationsaktivität.
Combining chemistry: The use of a lipase and a ruthenium catalyst allows the direct preparation of polysubstituted decalines with high optical and chemical yields from racemic alcohols (see scheme). The lipase-catalyzed kinetic resolution of the racemic alcohols, the ruthenium-catalyzed racemization of the slow-reacting enantiomers, and an intramolecular Diels–Alder reaction of the resultant esters all occur under identical conditions.
Enzymatische Unterstützung: Die Verwendung einer Lipase und eines Ruthenium-Katalysators ermöglicht die direkte Herstellung polysubstituierter Decaline aus racemischen Alkoholen mit hohem Enantiomerenüberschuss und in guten Ausbeuten (siehe Schema). Die Lipase-katalysierte kinetische Racematspaltung, die Ruthenium-katalysierte Racemisierung und eine intramolekulare Diels-Alder-Reaktion der resultierenden Ester laufen unter identischen Bedingungen ab.
A new method for obtaining optically pure 5-norbornene 2-endo-aldehyde derivatives was developed. The reaction of a diastereomeric mixture of the ene acetals 2 and 2′, derived from racemic norbornene aldehydes (±)-1 and chiral nonracemic (S,S)-hydrobenzoin 7, with NBS (0.5–0.6 eq.) in the presence of H2O proceeded in a kinetically controlled manner to give the optically pure hydroxy aldehydes 3 along with the intact ene acetals 2′. Both compounds 3 and 2′ were converted into the optically pure norbornene aldehydes 1 and ent-1, respectively. This method opens the way to produce various types of 5-norbornene 2-endo-aldehydes with 3-exo- or 3-endo-substituents in optically pure forms. Chirality 15:60–67, 2003. © 2002 Wiley-Liss, Inc.
The facile and efficient oxidation of various alcohols such as benzylic alcohols, primary alcohols, secondary alcohols, and diols in water using the hypervalent iodine(III) reagent, iodosobenzene (PhI=O), with KBr is described. Electrospray ionization (ESI) mass spectrometric studies on the behavior of PhI=O-KBr in aqueous solution suggested that these reactions are induced by the formation of highly reactive iodine species [PhI(Br)nO –]. Further development to recyclable polymer-supported iodine(III) reagent extends the utility of this reaction to afford an environmentally benign method.
The oxidative intramolecular coupling reaction of phenol ether derivatives (nonphenolic derivatives) on treatment with a novel combination of a hypervalent iodine(III) reagent, phenyliodine bis(trifluoroacetate) (PIFA), and heteropoly acid (HPA) was studied. Biaryl compounds were obtained in excellent yields on treatment of highly substituted phenol ethers. On the other hand, spirodienones were specifically formed when one of the preferred arylic coupling sites was substituted with a methoxy group in the para position.
A convenient method to determine the absolute configuration of trans-2-aryl cyclohexanols, 1-aryl alcohols and amines was achieved. This method takes advantage of the 1H NMR spectroscopic observations of the remarkable high-field shift of C18-CH3 protons caused by the aromatic shielding effect. It is based on a discrimination of the difference of the environments in two diastereomers derived from 3β-acetoxy-5-etienic acid. Furthermore, it was observed that the corresponding diastereomeric derivatives of the pyridyl alcohols were simply separated by extraction based on the difference in their basicity.
The first lipase-catalyzed domino reaction is described in which the acyl moiety formed during the enzymatic kinetic resolution of furfuryl alcohols (±)-3 with a 1-ethoxyvinyl ester 2 was utilized as a part of the constituent structure for the subsequent Diels–Alder reaction. The preparation of ester 2 from carboxylic acid 1 and the subsequent domino reaction were carried out in a one-pot reaction. Therefore, this procedure provides a convenient preparation of the optically active 7-oxabicyclo[2.2.1]heptene derivatives 5, which has five chiral, non-racemic carbon centers, from achiral 1 and racemic 3. The overall efficiency of this process was dependent on the substituent at the C-3 position of 3, and the use of the 3-methylfurfuryl derivatives, (±)-3 b and (±)-3 f, exclusively produced diastereoselectivity with excellent enantioselectivity to give (2R)-syn-5 (91–≥99 % ee) and (S)-3 (96–≥99 % ee). Similar procedures starting from the 3-bromofurfuryl alcohols (±)-3 h–j provided the cycloadducts (2R)-syn-5 j–q (93–≥99 % ee), in which the bromo group was utilized for the installation of bulky substituents to the 7-oxabicycloheptene core.
Eine elegante und effiziente Methode zum Aufbau der charakteristischen Schwefel-überbrückten Einheit der marinen antitumoralen Discorhabdin-Alkaloide wurde entwickelt. Durch Anwendung dieser Methode gelang die erste Totalsynthese von (±)-Discorhabdin A 1.
A concise and efficient method to construct the characteristic sulfur-cross-linked core of an unprecedented antitumor marine alkaloid, discorhabdins, has been developed. By utilizing this methodology the first total synthesis of (±)-discorhabdin A (1) has been achieved.
A linear approach to the total synthesis of racemic fredericamycin A (1) through the oxidative intramolecular [4+2] cycloaddition of a (phenylthio)acetylene–cobalt complex is described, which is applicable for the asymmetric total synthesis of naturally occuring 1. The highlight of this work is the aromatic Pummerer-type reaction with 1-ethoxyvinyl chloroacetate, which effects the introduction of the oxygen functional group to the internal B-ring of the highly functionalized, congested polyaromatic ABC-ring moiety.
17 Jahre nach der Isolierung des vielversprechenden Cytostaticums Fredericamycin A gelang seine asymmetrische Totalsynthese, womit auch die absolute Konfiguration geklärt werden konnte. Schlüsselschritt ist die regioselektive [4 + 2]-Cycloaddition von 3 an 2, das durch eine stereospezifische Umlagerung aus 1 erhalten wurde. Cp=(−)-Camphanoyl.
Seventeen years after the isolation of the promising antitumor antibiotic fredericamycin A, the first asymmetric total synthesis of this compound has been accomplished and thereby its absolute configuration established. The key feature is the regiocontrolled [4+2] cycloaddition of 3 to 2, which was obtained by the stereospecific rearrangement of 1. Cp = (−)-camphanoyl.