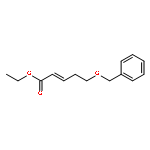
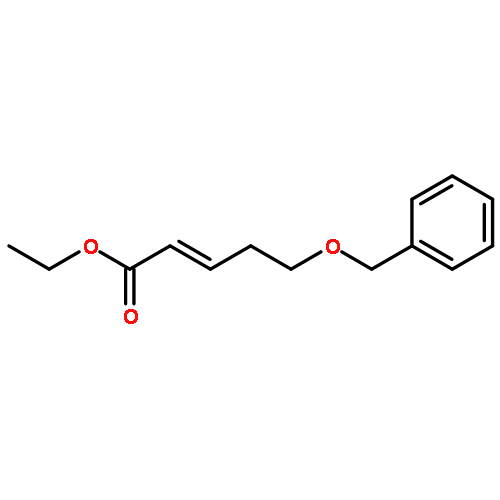
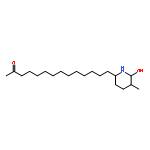
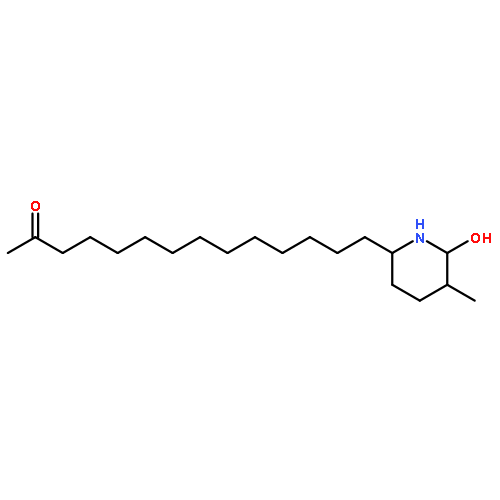
Co-reporter:Hiroyuki Konno, Takumi Onuma, Ikumi Nitanai, Masaki Wakabayashi, Shigekazu Yano, Kenta Teruya, Kenichi Akaji
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 12(Issue 12) pp:
Publication Date(Web):15 June 2017
DOI:10.1016/j.bmcl.2017.04.056
Synthesis and evaluation of new scaffold phenylisoserine derivatives connected with the essential functional groups against SARS CoV 3CL protease are described. The phenylisoserine backbone was found by simulation on GOLD software and the structure activity relationship study of phenylisoserine derivatives gave SK80 with an IC50 value of 43 μM against SARS CoV 3CL R188I mutant protease.Download high-res image (48KB)Download full-size image
Co-reporter:Hiroyuki Konno, Masaki Wakabayashi, Daiki Takanuma, Yota Saito, Kenichi Akaji
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 6) pp:1241-1254
Publication Date(Web):15 March 2016
DOI:10.1016/j.bmc.2016.01.052
Synthesis of serine derivatives having the essential functional groups for the inhibitor of SARS 3CL protease and evaluation of their inhibitory activities using SARS 3CL R188I mutant protease are described. The lead compounds, functionalized serine derivatives, were designed based on the tetrapeptide aldehyde and Bai’s cinnamoly inhibitor, and additionally performed with simulation on GOLD softwear. Structure activity relationship studies of the candidate compounds were given reasonable inhibitors ent-3 and ent-7k against SARS 3CL R188I mutant protease. These inhibitors showed protease selectivity and no cytotoxicity.
Co-reporter:Yasuhiro Shimamoto, Yasunao Hattori, Kazuya Kobayashi, Kenta Teruya, Akira Sanjoh, Atsushi Nakagawa, Eiki Yamashita, Kenichi Akaji
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 4) pp:876-890
Publication Date(Web):15 February 2015
DOI:10.1016/j.bmc.2014.12.028
The design and evaluation of a novel decahydroisoquinolin scaffold as an inhibitor for severe acute respiratory syndrome (SARS) chymotrypsin-like protease (3CLpro) are described. Focusing on hydrophobic interactions at the S2 site, the decahydroisoquinolin scaffold was designed by connecting the P2 site cyclohexyl group of the substrate-based inhibitor to the main-chain at the α-nitrogen atom of the P2 position via a methylene linker. Starting from a cyclohexene enantiomer obtained by salt resolution, trans-decahydroisoquinolin derivatives were synthesized. All decahydroisoquinolin inhibitors synthesized showed moderate but clear inhibitory activities for SARS 3CLpro, which confirmed the fused ring structure of the decahydroisoquinolin functions as a novel scaffold for SARS 3CLpro inhibitor. X-ray crystallographic analyses of the SARS 3CLpro in a complex with the decahydroisoquinolin inhibitor revealed the expected interactions at the S1 and S2 sites, as well as additional interactions at the N-substituent of the inhibitor.
Co-reporter:Yasunao Hattori, Kazuya Kobayashi, Ayaka Deguchi, Yukie Nohara, Tomomi Akiyama, Kenta Teruya, Akira Sanjoh, Atsushi Nakagawa, Eiki Yamashita, Kenichi Akaji
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 17) pp:5626-5640
Publication Date(Web):1 September 2015
DOI:10.1016/j.bmc.2015.07.023
A superior substrate sequence for BACE1 containing transition-state mimics at the scissile site was evaluated as a protease inhibitor. Hydroxymethylcarbonyl (HMC) and hydroxyethylamine (HEA) isosteres were selected as the transition state mimics, and incorporated into the scissile site of the superior sequence covering the P4 to P1’ sites (Glu-Ile-Thi-Thi*Nva; *denotes the cleavage site). Isosteres having different absolute configurations of the hydroxyl group were synthesized separately, and the effect of the configuration was evaluated. Configuration of the hydroxyl group of each isostere showed a marked effect on the inhibitory activity; anti-configuration to the scissile site substituent had potent inhibitory activity in an HMC-type inhibitor, whereas anti-configuration of HEA-type inhibitors showed no inhibitory activity. Structural evaluations based on X-ray crystallographic analyses of recombinant BACE1 in complex with each inhibitor provided insights into the protein–ligand interactions, especially at the prime sites.
Co-reporter:Chiyuki Awahara, Tadashi Tatsumi, Saki Furuta, Gen Shinjoh, Hiroyuki Konno, Kazuto Nosaka, Kazuya Kobayashi, Yasunao Hattori, Kenichi Akaji
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 8) pp:2482-2488
Publication Date(Web):15 April 2014
DOI:10.1016/j.bmc.2014.02.050
The effects of additional substituents covering the prime-site of retro-inverso (RI)-modified HTLV-1 protease inhibitors containing a hydroxyethylamine isoster were clarified. Stereo-selective construction of the most potent isoster backbone was achieved by the Evans-aldol reaction. Addition of N-acetylated d-amino acid corresponding to the P2′ site gave an RI-modified inhibitor showing superior inhibitory activity to the previous inhibitor. Inhibitory activities of the newly synthesized inhibitors suggest that partially modified RI inhibitors would interact with HTLV-1 protease in the same manner as the parent hydroxyethylamine inhibitor.
Co-reporter:Hiroyuki Konno, Hitoshi Endo, Satomi Ise, Keiki Miyazaki, Hideo Aoki, Akira Sanjoh, Kazuya Kobayashi, Yasunao Hattori, Kenichi Akaji
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 2) pp:685-690
Publication Date(Web):15 January 2014
DOI:10.1016/j.bmcl.2013.11.039
To research a new non-peptidyl inhibitor of beta-site amyloid precursor protein cleaving enzyme 1, we focused on the curcumin framework, two phenolic groups combined with an sp2 carbon spacer for low-molecular and high lipophilicity. The structure–activity relationship study of curcumin derivatives is described. Our results indicate that phenolic hydroxy groups and an alkenyl spacer are important structural factors for the inhibition of beta-site amyloid precursor protein cleaving enzyme 1 and, furthermore, non-competitive inhibition of enzyme activity is anticipated from an inhibitory kinetics experiment and docking simulation.
Co-reporter:Takaaki Mizuguchi, Naho Ohara, Mika Iida, Ryunosuke Ninomiya, Shinji Wada, Yoshiaki Kiso, Kazuki Saito, Kenichi Akaji
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 19) pp:5730-5737
Publication Date(Web):1 October 2012
DOI:10.1016/j.bmc.2012.08.013
Structure–activity relationships of cyclic peptides mimicking the β-hairpin structure of the ‘dimerization arm’ at residues 242–259 of the EGF receptor are examined. Cyclic peptides containing the arm head of the β-hairpin loop showed inhibitory activity toward the EGF receptor’s dimerization. Cyclic peptides containing a Retro-Inverso sequence of the dimerization arm showed clear inhibitory effects on the dimerization in vitro and efficiently suppressed the proliferation of A431 cells, which abundantly express the EGF receptor on their surface. The effects at a specific hydrophobic site of the loop structure were expected to enhance the interactions with the receptor.
Co-reporter:Kenichi Akaji ; Hiroyuki Konno ; Hironori Mitsui ; Kenta Teruya ; Yasuhiro Shimamoto ; Yasunao Hattori ; Takeshi Ozaki ; Masami Kusunoki ;Akira Sanjoh
Journal of Medicinal Chemistry 2011 Volume 54(Issue 23) pp:7962-7973
Publication Date(Web):October 20, 2011
DOI:10.1021/jm200870n
The design and evaluation of low molecular weight peptide-based severe acute respiratory syndrome (SARS) chymotrypsin-like protease (3CL) protease inhibitors are described. A substrate-based peptide aldehyde was selected as a starting compound, and optimum side-chain structures were determined, based on a comparison of inhibitory activities with Michael type inhibitors. For the efficient screening of peptide aldehydes containing a specific C-terminal residue, a new approach employing thioacetal to aldehyde conversion mediated by N-bromosuccinimide was devised. Structural optimization was carried out based on X-ray crystallographic analyses of the R188I SARS 3CL protease in a complex with each inhibitor to provide a tetrapeptide aldehyde with an IC50 value of 98 nM. The resulting compound carried no substrate sequence, except for a P3 site directed toward the outside of the protease. X-ray crystallography provided insights into the protein–ligand interactions.

![Benzenebutanoic acid, b-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-a-hydroxy-, (aS,bS)-](http://img.cochemist.com/ccimg/210800/210754-59-9.png)
![Benzenebutanoic acid, b-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-a-hydroxy-, (aS,bS)-](http://img.cochemist.com/ccimg/210800/210754-59-9_b.png)


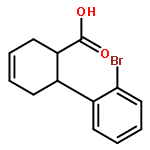
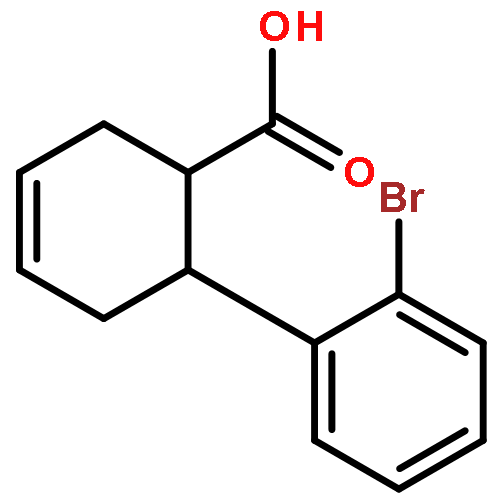
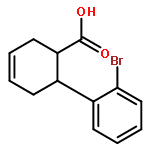
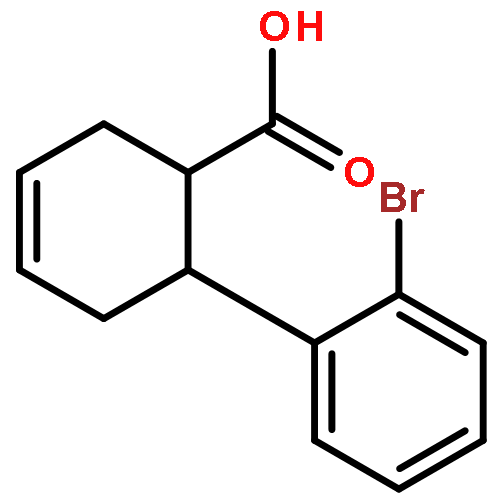



![L-SERINE, N-[(2-PROPENYLOXY)CARBONYL]-](http://img.cochemist.com/ccimg/90600/90508-22-8.png)
![L-SERINE, N-[(2-PROPENYLOXY)CARBONYL]-](http://img.cochemist.com/ccimg/90600/90508-22-8_b.png)
![CARBAMIC ACID, [(1S)-1-FORMYL-3-METHYLBUTYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/71500/71413-15-5.png)
![CARBAMIC ACID, [(1S)-1-FORMYL-3-METHYLBUTYL]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/71500/71413-15-5_b.png)