Co-reporter:Doo-Hyun Kwon, Matthew Proctor, Sergio Mendoza, Christopher Uyeda, and Daniel H. Ess
ACS Catalysis July 7, 2017 Volume 7(Issue 7) pp:4796-4796
Publication Date(Web):June 1, 2017
DOI:10.1021/acscatal.7b00978
Homodinuclear transition-metal catalysts with a direct metal–metal bond have the potential to induce novel reaction mechanisms and selectivity compared with mononuclear catalysts. The dinuclear (i-PrNDI)Ni2(C6H6) (NDI = naphthyridine-diimine) complex catalyzes selective cyclotrimerization of monosubstituted alkynes, whereas mononuclear Ni catalysts generally give cyclotetramerization of alkynes. Density functional theory calculations reveal that the homodinuclear Ni–Ni catalyst induces a spin crossover mechanism that involves metallacyclopentadiene and nonclassical bridging metallacycloheptatriene intermediates. The cis configuration of the nonclassical bridging metallacycloheptatriene Ni–vinyl bonds results in alkyne cyclotrimerization by fast reductive elimination. This dinuclear mechanism differs from previously reported mononuclear Ni mechanisms and provides an explanation for cyclotrimerization versus cyclotetramerization selectivity and arene regioselectivity.Keywords: alkyne cyclotrimerization; catalysis; density functional theory; dinuclear; nickel;
Co-reporter:Padmanabha V. Kattamuri, Jun Yin, Surached Siriwongsup, Doo-Hyun Kwon, Daniel H. Ess, Qun Li, Guigen Li, Muhammed Yousufuddin, Paul F. Richardson, Scott C. Sutton, and László Kürti
Journal of the American Chemical Society August 16, 2017 Volume 139(Issue 32) pp:11184-11184
Publication Date(Web):June 26, 2017
DOI:10.1021/jacs.7b05279
Given the importance of amines in a large number of biologically active natural products, active pharmaceutical ingredients, agrochemicals, and functional materials, the development of efficient C–N bond-forming methods with wide substrate scope continues to be at the frontier of research in synthetic organic chemistry. Here, we present a general and fundamentally new synthetic approach for the direct, transition-metal-free preparation of symmetrical and unsymmetrical diaryl-, arylalkyl-, and dialkylamines that relies on the facile single or double addition of readily available C-nucleophiles to the nitrogen atom of bench-stable electrophilic aminating agents. Practical single and double polarity reversal (i.e., umpolung) of the nitrogen atom is achieved using sterically and electronically tunable ketomalonate-derived imines and oximes. Overall, this novel approach represents an operationally simple, scalable, and environmentally friendly alternative to transition-metal-catalyzed C–N cross-coupling methods that are currently used to access structurally diverse secondary amines.
Co-reporter:Colin M. Kelly;Jack T. Fuller III;Casper M. Macaulay;Dr. Robert McDonald;Dr. Michael J. Ferguson;Dr. Steven M. Bischof;Dr. Orson L. Sydora; Dr. Daniel H. Ess; Dr. Mark Stradiotto; Dr. Laura Turculet
Angewandte Chemie 2017 Volume 129(Issue 22) pp:6409-6413
Publication Date(Web):2017/05/22
DOI:10.1002/ange.201700857
AbstractThe first examples of stoichiometric dehydrogenative B−H/C(sp3)−H benzylic borylation reactions, which are of relevance to catalytic methylarene (di)borylation, are reported. These unusual transformations involving a (κ2-P,N)Pt(η3-benzyl) complex, and either pinacolborane or catecholborane, proceed cleanly at room temperature. Density functional calculations suggest that borylation occurs via successive σ-bond metathesis steps, whereby a PtII−H intermediate engages in C(sp3)−H bond activation-induced dehydrogenation.
Co-reporter:Clinton R. King, Samantha J. Gustafson, Benjamin R. Black, Steven K. Butler, Michael M. Konnick, Roy A. Periana, Brian M. Hashiguchi, and Daniel H. Ess
Organometallics 2017 Volume 36(Issue 1) pp:109-113
Publication Date(Web):August 25, 2016
DOI:10.1021/acs.organomet.6b00475
M06 density functional theory calculations reveal that arene C–H functionalization by the p-block main-group-metal complex TlIII(TFA)3 (TFA = trifluoroacetate) occurs by a C–H activation mechanism akin to transition-metal-mediated C–H activation. For benzene, toluene, and xylenes a one-step C–H activation is preferred over electron transfer or proton-coupled electron transfer. The proposed C–H activation mechanism is consistent with calculation and comparison to experiment, of arene thallation rates, regioselectivity, and H/D kinetic isotope effects. For tetramethyl- and pentamethyl-substituted arenes, electron transfer becomes a competitive pathway and thermodynamic and kinetic calculations correctly predict the experimentally reported electron transfer crossover region. These calculations show that p-block metals activate strong hydrocarbon C–H bonds through organometallic intermediates and changes in arene functional groups can result in a shift from C–H activation to electron transfer.
Co-reporter:Jack T. Fuller III, Steven Butler, Deepa Devarajan, Austin Jacobs, Brian G. Hashiguchi, Michael M. Konnick, William A. Goddard III, Jason Gonzales, Roy A. Periana, and Daniel H. Ess
ACS Catalysis 2016 Volume 6(Issue 7) pp:4312
Publication Date(Web):June 8, 2016
DOI:10.1021/acscatal.6b00226
Methane conversion to methyl bisulfate by HgII(SO4) in sulfuric acid is an example of fast and selective alkane oxidation catalysis. Dichotomous mechanisms involving C–H activation and electron transfer have been proposed based on experiments. Radical oxidation pathways have also been proposed for some reaction conditions. HgII is also of significant interest because as a d10 transition metal it is similar to d10 main-group metals that also oxidize alkanes. Density-functional calculations are presented that use both implicit and a mixture of implicit/explicit solvent models for the complete HgII catalytic cycle of methane oxidation to methyl bisulfate. These calculations are consistent with experiment and reveal that methane is functionalized to methyl bisulfate by a C–H activation and reductive metal alkyl functionalization mechanism. This reaction pathway is lower in energy than both electron transfer and proton-coupled electron transfer pathways. After methane C–H functionalization, catalysis is completed by conversion of the proposed resting state, [HgI(HSO4)]2, into Hg0 followed by Hg0 to HgII oxidation induced by SO3 from dehydration of sulfuric acid. This catalytic cycle is efficient because in sulfuric acid the HgII/Hg0 potential results in a moderate free energy barrier for oxidation (∼40 kcal/mol) and HgII is electrophilic enough to induce barriers of <40 kcal/mol for C–H activation and reductive metal alkyl functionalization. Comparison of HgII to TlIII shows that while C–H activation and reductive metal alkyl functionalization have reasonable barriers for TlIII, the oxidation of TlI to TlIII has a significantly larger barrier than Hg0 to HgII oxidation and therefore TlIII is not catalytic in sulfuric acid. Comparison of HgII to CdII and ZnII reveals that while M0 to MII oxidation and C–H activation are feasible for these first-row and second-row transition metals, reductive metal alkyl functionalization barriers are very large and catalysis is not feasible. Calculations are also presented that outline the mechanism and energy landscape for radical-initiated (K2S2O8) methane oxidation to methanesulfonic acid in sulfuric acid.Keywords: C−H activation; density functional theory; electron transfer; mercury; metal alkyl functionalization; methane; proton-coupled electron transfer; radical initiation; sulfuric acid; thallium
Co-reporter:Ryan W. Carlsen and Daniel H. Ess
Dalton Transactions 2016 vol. 45(Issue 24) pp:9835-9840
Publication Date(Web):19 Feb 2016
DOI:10.1039/C6DT00256K
Transition metal heterobimetallic complexes with dative metal–metal interactions have the potential for novel fast reactivity. There are few studies that both compare the reactivity of different metal centers in heterobimetallic complexes and compare bimetallic reactivity to monometallic reactivity. Here we report density-functional calculations that show the reactivity of [Cl2Ti(NtBuPPh2)2MII(η3-methallyl)] heterobimetallic complexes for allylic amination follows M = Ni > Pd > Pt. This reactivity trend was not anticipated since the amine addition transition state involves MII to M0 reduction and this could disadvantage Ni. Comparison of heterobimetallic complexes to the corresponding monometallic (CH2)2(NtBuPPh2)2MII(η3-methallyl) complexes reveals that this reactivity trend is due to the bimetallic interaction and that the bimetallic interaction significantly lowers the barrier height for amine addition by >10 kcal mol−1. The impact of the early transition metal center on the amination addition barrier height depends on the late transition metal center. The lowest barrier heights for this reaction occur when late and early transition metal centers are from the same periodic table row.
Co-reporter:Whitney K. Walker; Benjamin M. Kay; Scott A. Michaelis; Diana L. Anderson; Stacey J. Smith; Daniel H. Ess;David J. Michaelis
Journal of the American Chemical Society 2015 Volume 137(Issue 23) pp:7371-7378
Publication Date(Web):May 6, 2015
DOI:10.1021/jacs.5b02428
Experiments and density functional calculations were used to quantify the impact of the Pd–Ti interaction in the cationic heterobimetallic Cl2Ti(NtBuPPh2)2Pd(η3-methallyl) catalyst 1 used for allylic aminations. The catalytic significance of the Pd–Ti interaction was evaluated computationally by examining the catalytic cycle for catalyst 1 with a conformation where the Pd–Ti interaction is intact versus one where the Pd–Ti interaction is severed. Studies were also performed on the relative reactivity of the cationic monometallic (CH2)2(NtBuPPh2)2Pd(η3-methallyl) catalyst 2 where the Ti from catalyst 1 was replaced by an ethylene group. These computational and experimental studies revealed that the Pd–Ti interaction lowers the activation barrier for turnover-limiting amine reductive addition and accelerates catalysis up to 105. The Pd–Ti distance in 1 is the result of the NtBu groups enforcing a boat conformation that brings the two metals into close proximity, especially in the transition state. The turnover frequency of classic Pd π allyl complexes was compared to that of 1 to determine the impact of P–Pd–P coordination angle and ligand electronic properties on catalysis. These experiments identified that cationic (PPh3)2Pd(η3-CH2C(CH3)CH2) catalyst 3 performs similarly to 1 for allylic aminations with diethylamine. However, computations and experiment reveal that the apparent similarity in reactivity is due to very fast reaction kinetics. The higher reactivity of 1 versus 3 was confirmed in the reaction of methallyl chloride and 2,2,6,6-tetramethylpiperidine (TMP). Overall, experiments and calculations demonstrate that the Pd–Ti interaction induces and is responsible for significantly lower barriers and faster catalysis for allylic aminations.
Co-reporter:Ying Zhang, Samuel P. Roberts, Robert G. Bergman, and Daniel H. Ess
ACS Catalysis 2015 Volume 5(Issue 3) pp:1840
Publication Date(Web):February 20, 2015
DOI:10.1021/cs501884j
Transition metal heterobimetallic catalysts provide an alternative to classic transition metal ligand catalyst design. The resurgence in popularity of heterobimetallic complexes prompted our use of density functional theory to examine the mechanism and reactivity of alkene hydrogenation catalyzed by the transition metal heterobimetallic complex Cp2Ta(CH2)2Ir(CO)(PPh3) and the transition metal/main group complex Ph2P(CH2)2Ir(CO)(PPh3). Calculations indicate that the Ir–Ta and Ir–P catalysts operate by different mechanisms. For the Ir–Ta complex, initial H2 oxidative addition to the Ir metal center followed by reductive elimination of an Ir–H and μ-CH2 bridge transforms the starting heterobimetallic complex into an active Ir–H catalyst. This catalyst precursor transformation occurs because the cationic Cp2Ta group provides a low activation barrier for reductive elimination. This transformation does not occur for the Ir–P catalyst because the reductive elimination activation barrier is significantly higher in energy. The active heterobimetallic Ir–H likely catalyzes multiple turnovers of alkene hydrogenation before reforming the original heterobimetallic Ir–Ta complex. The Ir–H catalytic cycle involves a series of classic organometallic reaction steps: alkene migratory insertion, H2 oxidative addition, and reductive elimination. In the Ir–P mechanism, the Ph2P(CH2)2 group remains as a spectator ligand throughout the active catalytic cycle. The Ir–P catalytic cycle involves H2 oxidative addition, phosphine ligand dissociation, ethylene migratory insertion, and reductive elimination.Keywords: alkene hydrogenation; catalysis; density functional theory; heterobimetallic; iridium; mechanism; tantalum
Co-reporter:Deepa Devarajan; Samantha J. Gustafson; F. Matthias Bickelhaupt
Journal of Chemical Education 2015 Volume 92(Issue 2) pp:286-290
Publication Date(Web):November 19, 2014
DOI:10.1021/ed5005905
Undergraduate organic chemistry textbooks and Internet websites use a variety of approaches for presenting and explaining the impact of halogen atom size on trends in bond strengths and/or acidity of hydrogen halides. In particular, several textbooks and Internet websites explain these trends by invoking decreasing orbital overlap between the hydrogen 1s atomic orbital and successively larger group 17 halogen atomic orbitals. A similar orbital overlap rationalization is often extended to the trends in alkyl halide bond strengths. We examined this orbital overlap explanation using quantum mechanical calculations. Calculations reveal that orbital overlap increases rather than decreases with successively larger group 17 halogen atomic orbitals. This suggests that an orbital overlap explanation is physically incorrect and unneeded. Alternative to orbital overlap, we briefly discuss physically correct models for rationalizing halogen bond strength and acidity based on quantum mechanical valence bond theory and molecular orbital theory.
Co-reporter:Austin Talbot, Deepa Devarajan, Samantha J. Gustafson, Israel Fernández, F. Matthias Bickelhaupt, and Daniel H. Ess
The Journal of Organic Chemistry 2015 Volume 80(Issue 1) pp:548-558
Publication Date(Web):December 4, 2014
DOI:10.1021/jo5025514
Heteroaromatic azadienes, especially 1,2,4,5-tetrazines, are extremely reactive partners with alkenes in inverse-electron-demand Diels–Alder reactions. Azadiene cycloaddition reactions are used to construct heterocycles in synthesis and are popular as bioorthogonal reactions. The origin of fast azadiene cycloaddition reactivity is classically attributed to the inverse frontier molecular orbital (FMO) interaction between the azadiene LUMO and alkene HOMO. Here, we use a combination of ab initio, density functional theory, and activation-strain model calculations to analyze physical interactions in heteroaromatic azadiene–alkene cycloaddition transition states. We find that FMO interactions do not control reactivity because, while the inverse FMO interaction becomes more stabilizing, there is a decrease in the forward FMO interaction that is offsetting. Rather, fast cycloadditions are due to a decrease in closed-shell Pauli repulsion between cycloaddition partners. The kinetic–thermodynamic relationship found for these inverse-electron-demand cycloadditions is also due to the trend in closed-shell repulsion in the cycloadducts. Cycloaddition regioselectivity, however, is the result of differences in occupied–unoccupied orbital interactions due to orbital overlap. These results provide a new predictive model and correct physical basis for heteroaromatic azadiene reactivity and regioselectivity with alkene dieneophiles.
Co-reporter:Samantha J. Gustafson, Jack T. Fuller III, Deepa Devarajan, Justin Snyder, Roy A. Periana, Brian G. Hashiguchi, Michael M. Konnick, and Daniel H. Ess
Organometallics 2015 Volume 34(Issue 22) pp:5485-5495
Publication Date(Web):November 10, 2015
DOI:10.1021/acs.organomet.5b00849
Activation and functionalization of alkane C–H bonds has historically been dominated by transition-metal complexes. Light alkanes can also be partially oxidized by sixth-row main-group compounds, such as TlIII(TFA)3 (TFA = trifluoroacetate). Here we present density-functional calculations which demonstrate that TlIII(TFA)3 oxidizes alkanes by closed-shell C–H activation and Tl–alkyl functionalization mechanisms. The discovery of a C–H activation pathway is surprising, because TlIII often oxidizes arene C–H bonds through an electron transfer mechanism and the transition-metal complex CoIII(TFA)3, with similar oxidation state and ligand coordination, oxidizes alkanes via an open-shell radical mechanism. Comparison of TlIII(TFA)3 to the transition-metal analogue IrIII(TFA)3 reveals that key to TlIII oxidation of alkanes is a moderate barrier for C–H bond activation that is lower in energy than open-shell pathways and a subsequent metal–alkyl functionalization reaction step with a very low barrier. Our calculations suggest that the high-spin ground state of CoIII(TFA)3 provides a low-energy open-shell decarboxylation pathway that leads to radical oxidation of alkanes, which is not available for the d10 TlIII(TFA)3 complex. The C–H activation pathway and transition state model provide a straightforward explanation for why TlIII(TFA)3 promotes alkane C–H bond activation but HgII(TFA)2 does not.
Co-reporter:Jack T. Fuller, Daniel J. Harrison, Matthew C. Leclerc, R. Tom Baker, Daniel H. Ess, and Russell P. Hughes
Organometallics 2015 Volume 34(Issue 21) pp:5210-5213
Publication Date(Web):October 26, 2015
DOI:10.1021/acs.organomet.5b00863
Density functional calculations (M06/def2-TZVP//M06/LACVP** in THF solvent) reveal that the formation of a perfluorometallacyclobutane from tetrafluoroethylene, C2F4 (TFE), and the difluorocarbene complex CoCp(PPh2Me)(CF2) is highly exothermic and involves a stepwise [2 + 2] cycloaddition mechanism with a 1,4-singlet diradical intermediate. This pathway is significantly lower in energy than pathways involving initial η1 or η2 binding of TFE to cobalt. The stability of the singlet-diradical intermediate results from the formation of a strong CF2–CF2 bond coupled with the radical stabilizing effect of a difluoromethylene group. A concerted [2 + 2] transition state between the 18-electron complex CoCp(PPh2Me)(CF2) and TFE is very high in energy and essentially forbidden.
Co-reporter:Sean M. McCarthy ; Yi-Chun Lin ; Deepa Devarajan ; Ji Woong Chang ; Hemant P. Yennawar ; Robert M. Rioux ; Daniel H. Ess ;Alexander T. Radosevich
Journal of the American Chemical Society 2014 Volume 136(Issue 12) pp:4640-4650
Publication Date(Web):March 5, 2014
DOI:10.1021/ja412469e
Ammonia, alkyl amines, and aryl amines are found to undergo rapid intermolecular N–H oxidative addition to a planar mononuclear σ3-phosphorus compound (1). The pentacoordinate phosphorane products (1·[H][NHR]) are structurally robust, permitting full characterization by multinuclear NMR spectroscopy and single-crystal X-ray diffraction. Isothermal titration calorimetry was employed to quantify the enthalpy of the N–H oxidative addition of n-propylamine to 1 (nPrNH2 + 1 → 1·[H][NHnPr], ΔHrxn298 = −10.6 kcal/mol). The kinetics of n-propylamine N–H oxidative addition were monitored by in situ UV absorption spectroscopy and determination of the rate law showed an unusually large molecularity (ν = k[1][nPrNH2]3). Kinetic experiments conducted over the temperature range of 10–70 °C revealed that the reaction rate decreased with increasing temperature. Activation parameters extracted from an Eyring analysis (ΔH⧧ = −0.8 ± 0.4 kcal/mol, ΔS⧧ = −72 ± 2 cal/(mol·K)) indicate that the cleavage of strong N–H bonds by 1 is entropy controlled due to a highly ordered, high molecularity transition state. Density functional calculations indicate that a concerted oxidative addition via a classical three-center transition structure is energetically inaccessible. Rather, a stepwise heterolytic pathway is preferred, proceeding by initial amine-assisted N–H heterolysis upon complexation to the electrophilic phosphorus center followed by rate-controlling N → P proton transfer.
Co-reporter:Michael M. Konnick ; Steven M. Bischof ; Muhammed Yousufuddin ; Brian G. Hashiguchi ; Daniel H. Ess ;Roy A. Periana
Journal of the American Chemical Society 2014 Volume 136(Issue 28) pp:10085-10094
Publication Date(Web):June 13, 2014
DOI:10.1021/ja504368r
The selective, oxidative functionalization of ethane, a significant component of shale gas, to products such as ethylene or ethanol at low temperatures and pressures remains a significant challenge. Herein we report that ethane is efficiently and selectively functionalized to the ethanol ester of H2SO4, ethyl bisulfate (EtOSO3H) as the initial product, with the PtII “Periana-Catalytica” catalyst in 98% sulfuric acid. A subsequent organic reaction selectively generates isethionic acid bisulfate ester (HO3S-CH2-CH2-OSO3H, ITA). In contrast to the modest 3–5 times faster rate typically observed in electrophilic CH activation of higher alkanes, ethane CH functionalization was found to be ∼100 times faster than that of methane. Experiment and quantum-mechanical calculations reveal that this unexpectedly large increase in rate is the result of a fundamentally different catalytic cycle in which ethane CH activation (and not platinum oxidation as for methane) is now turnover limiting. Facile PtII-Et functionalization was determined to occur via a low energy β-hydride elimination pathway (which is not available for methane) to generate ethylene and a PtII-hydride, which is then rapidly oxidized by H2SO4 to regenerate PtII-X2. A rapid, non-Pt-catalyzed reaction of formed ethylene with the hot, concentrated H2SO4 solvent cleanly generate EtOSO3H as the initial product, which further reacts with the H2SO4 solvent to generate ITA.
Co-reporter:Zhiwei Ma, Jintao Jiang, Shi Luo, Yu Cai, Joseph M. Cardon, Benjamin M. Kay, Daniel H. Ess, and Steven L. Castle
Organic Letters 2014 Volume 16(Issue 15) pp:4044-4047
Publication Date(Web):July 16, 2014
DOI:10.1021/ol5018933
A concise synthesis of peptides that contain E- or Z-dehydroisoleucine (ΔIle) residues is reported. The key reaction is an unusual anti dehydration of β-tert-hydroxy amino acid derivatives that is mediated by the Martin sulfurane. A subsequent tandem Staudinger reduction–O → N acyl transfer process forges an amide bond to the ΔIle residue with minimal E/Z alkene isomerization. Density functional calculations attribute the stereospecific dehydration to a highly asynchronous E2 anti process.
Co-reporter:Samantha A. Burgess, Deepa Devarajan, Tamara Bolaño, Daniel H. Ess, T. Brent Gunnoe, Michal Sabat, and William H. Myers
Inorganic Chemistry 2014 Volume 53(Issue 10) pp:5328-5340
Publication Date(Web):May 7, 2014
DOI:10.1021/ic500636m
The Rh(III) complexes [(tbpy)2Rh(OMe)(L)][X]n (tbpy = 4,4′-di-tert-butyl-2,2′-bipyridyl; L = MeOH, n = 2, X = OTf (OTf = trifluoromethanesulfonate), TFA (TFA = trifluoroacetate); L = TFA, n = 1, X = OTf) have been shown to activate dihydrogen via net 1,2-addition of the H–H bond across the RhIII–OMe bond. The bis(methoxide) complex [(tbpy)2Rh(OMe)2][OTf] was synthesized by addition of CsOH·H2O in methanol to [(tbpy)2Rh(OTf)2][OTf] in CH3CN. The addition of HTFA to [(tbpy)2Rh(OMe)2][OTf] leads to the formation of [(tbpy)2Rh(OMe)(MeOH)][OTf][TFA], which exists in equilibrium with [(tbpy)2Rh(OMe)(TFA)][OTf]. The mixture of [(tbpy)2Rh(OMe)(MeOH)][OTf][TFA] and [(tbpy)2Rh(OMe)(TFA)][OTf] activates dihydrogen at 68 °C to give methanol and [(tbpy)2Rh(H)(TFA)][OTf]. Studies indicate that the activation of dihydrogen has a first-order dependence on the Rh(III) methoxide complex and a dependence on hydrogen that is between zero and first order. Combined experimental and computational studies have led to a proposed mechanism for hydrogen activation by [(tbpy)2Rh(OMe)(MeOH)][OTf][TFA] that involves dissociation of MeOH, coordination of hydrogen, and 1,2-addition of hydrogen across the Rh–OMe bond. DFT calculations indicate that there is a substantial energy penalty for MeOH dissociation and a relatively flat energy surface for subsequent hydrogen coordination and activation.
Co-reporter:Brian G. Hashiguchi;Michael M. Konnick;Steven M. Bischof;Samantha J. Gustafson;Deepa Devarajan;Niles Gunsalus;Roy A. Periana
Science 2014 Volume 343(Issue 6176) pp:1232-1237
Publication Date(Web):14 Mar 2014
DOI:10.1126/science.1249357
Light Alkanes, Heavy Metals
Hydraulic fracturing, or fracking, has rapidly increased the supply of natural gas and has motivated methods to convert its constituents into commodity chemicals. Hashiguchi et al. (p. 1232) have found that lead and thallium salts are both efficient and selective oxidants, not only for methane, but for ethane and propane as well. In trifluoroacetic acid solvent, the alkanes are cleanly oxidized to the trifluoroacetate esters of their respective alcohols and 1,2-diols. Building on earlier discoveries, this work paves the way to developing methods that reduce our dependence on petroleum for industrial feedstocks.
Co-reporter:Jawahar L. Jat;Mahesh P. Paudyal;Hongyin Gao;Qing-Long Xu;Muhammed Yousufuddin;Deepa Devarajan;László Kürti;John R. Falck
Science 2014 Volume 343(Issue 6166) pp:61-65
Publication Date(Web):03 Jan 2014
DOI:10.1126/science.1245727
Unadorned Aziridines
Multiple catalytic methods have been developed to make aziridines—strained triangular carbon-nitrogen-carbon rings that function as versatile synthetic intermediates. However, the majority require protection of the nitrogen precursor with a sulfonyl group that is subsequently inconvenient to remove. Jat et al. (p. 61; see the Perspective by Türkmen and Aggarwal) used a hydroxylamine derivative as the nitrogen source together with an established rhodium catalyst to prepare a wide range of unprotected aziridines, with nitrogen bonded simply to hydrogen or a methyl group.
Co-reporter:Dr. Hongyin Gao;Dr. Qing-Long Xu;Dr. Muhammed Yousufuddin; Daniel H. Ess; László Kürti
Angewandte Chemie International Edition 2014 Volume 53( Issue 10) pp:2701-2705
Publication Date(Web):
DOI:10.1002/anie.201309973
Abstract
We disclose an efficient and operationally simple protocol for the preparation of fused N-heterocycles starting from readily available 2-nitrobiaryls and PhMgBr under mild conditions. More than two dozen N-heterocycles, including two bioactive natural products, have been synthesized using this method. A stepwise electrophilic aromatic cyclization mechanism was proposed by DFT calculations.
Co-reporter:Peter Anderson, Alban Petit, Junming Ho, Mariusz Pawel Mitoraj, Michelle L. Coote, David Danovich, Sason Shaik, Benoît Braïda, and Daniel H. Ess
The Journal of Organic Chemistry 2014 Volume 79(Issue 21) pp:9998-10001
Publication Date(Web):October 15, 2014
DOI:10.1021/jo501549q
Accurate gas-phase and solution-phase valence bond calculations reveal that protonation of the hydroxyl group of aliphatic alcohols transforms the C–O bond from a principally covalent bond to a complete charge-shift bond with principally “no-bond” character. All bonding in this charge-shift bond is due to resonance between covalent and ionic structures, which is a different bonding mechanism from that of traditional covalent bonds. Until now, charge-shift bonds have been previously identified in inorganic compounds or in exotic organic compounds. This work showcases that charge-shift bonds can occur in common organic species.
Co-reporter:Dr. Hongyin Gao;Dr. Qing-Long Xu;Dr. Muhammed Yousufuddin; Daniel H. Ess; László Kürti
Angewandte Chemie 2014 Volume 126( Issue 10) pp:2739-2743
Publication Date(Web):
DOI:10.1002/ange.201309973
Abstract
We disclose an efficient and operationally simple protocol for the preparation of fused N-heterocycles starting from readily available 2-nitrobiaryls and PhMgBr under mild conditions. More than two dozen N-heterocycles, including two bioactive natural products, have been synthesized using this method. A stepwise electrophilic aromatic cyclization mechanism was proposed by DFT calculations.
Co-reporter:Qing-Long Xu ; Hongyin Gao ; Muhammed Yousufuddin ; Daniel H. Ess ;László Kürti
Journal of the American Chemical Society 2013 Volume 135(Issue 38) pp:14048-14051
Publication Date(Web):September 4, 2013
DOI:10.1021/ja4074563
We disclose a facile, aerobic, transition-metal-free, direct, and regiospecific mono-α-arylation of ketones to yield aryl benzyl and (cyclo)alkyl benzyl ketones with substitution patterns that are currently inaccessible or challenging to prepare using conventional methods. The transformation is operationally simple, scalable, and environmentally friendly. There is no need for pre-functionalization (i.e., α-halogenation or silyl enol ether formation) or the use of specialized arylating agents (i.e., diaryliodonium salts). DFT calculations suggest that the in situ-generated enolate undergoes direct C–C bond formation with the nitroarene followed by regioselective O2-mediated C–H oxidation.
Co-reporter:Ulrich Hintermair ; Stafford W. Sheehan ; Alexander R. Parent ; Daniel H. Ess ; David T. Richens ; Patrick H. Vaccaro ; Gary W. Brudvig ;Robert H. Crabtree
Journal of the American Chemical Society 2013 Volume 135(Issue 29) pp:10837-10851
Publication Date(Web):July 3, 2013
DOI:10.1021/ja4048762
We present evidence for Cp* being a sacrificial placeholder ligand in the [Cp*IrIII(chelate)X] series of homogeneous oxidation catalysts. UV–vis and 1H NMR profiles as well as MALDI-MS data show a rapid and irreversible loss of the Cp* ligand under reaction conditions, which likely proceeds through an intramolecular inner-sphere oxidation pathway reminiscent of the reductive in situ elimination of diolefin placeholder ligands in hydrogenation catalysis by [(diene)MI(L,L′)]+ (M = Rh and Ir) precursors. When oxidatively stable chelate ligands are bound to the iridium in addition to the Cp*, the oxidized precursors yield homogeneous solutions with a characteristic blue color that remain active in both water- and CH-oxidation catalysis without further induction period. Electrophoresis suggests the presence of well-defined Ir-cations, and TEM-EDX, XPS, 17O NMR, and resonance-Raman spectroscopy data are most consistent with the molecular identity of the blue species to be a bis-μ-oxo di-iridium(IV) coordination compound with two waters and one chelate ligand bound to each metal. DFT calculations give insight into the electronic structure of this catalyst resting state, and time-dependent simulations agree with the assignments of the experimental spectroscopic data. [(cod)IrI(chelate)] precursors bearing the same chelate ligands are shown to be equally effective precatalysts for both water- and CH-oxidations using NaIO4 as chemical oxidant.
Co-reporter:Deepa Devarajan, Charles E. Doubleday, and Daniel H. Ess
Inorganic Chemistry 2013 Volume 52(Issue 15) pp:8820-8833
Publication Date(Web):July 9, 2013
DOI:10.1021/ic4010399
Density functional theory (DFT), absolutely localized molecular orbital (ALMO) analysis, and quasiclassical trajectories (QCTs) were used to study the structure, barrier heights, thermodynamics, electronic properties, and dynamics of dihydrogen (H2) activation by singlet divalent main group compounds (ER2; E = C, Si, Ge). ALMO energy and charge decomposition calculations reveal that in the transition state CR2 acts as an ambiphile toward H2 because of equal forward-bonding and back-bonding orbital stabilization while SiR2 and GeR2 act as nucleophiles with dominant orbital energy stabilization arising from ER2 to H2 donation. Frontier molecular orbital (FMO) energy gaps do not provide a reasonable estimate of energy stabilization gained between the ER2 and H2 in the transition state or an accurate description of the nucleophilic versus electrophilic character because of electron repulsion and orbital overlap influences that are neglected. In CR2 transition states, forward-bonding and back-bonding are maximized in the nonleast motion geometry. In contrast, SiR2/GeR2 transition states have side-on geometries to avoid electron–electron repulsion. Electron repulsion, rather than orbital interactions, also determines the relative barrier heights of CR2 versus SiR2/GeR2 reactions. Examination of barrier heights and reaction energies shows a clear kinetic-thermodynamic relationship for ER2 activation of H2. A computational survey of R groups on ER2 divalent atom centers was performed to explore the possibility for H2 activation to occur with a low barrier and thermodynamically reversible. QCTs show that dihydrogen approach and reaction with CR2 may involve geometries significantly different than the static transition-state structure. In contrast, trajectories for dihydrogen addition to SiR2 involve geometries close to the side-on approach suggested by the static transition-state structure. QCTs also demonstrate that addition of H2 to CR2 and SiR2 is dynamically concerted with the average time gap of bond formation between E–H bonds of approximately 11 and 21 fs, respectively.
Co-reporter:Steven E. Kalman, Alban Petit, T. Brent Gunnoe, Daniel H. Ess, Thomas R. Cundari, and Michal Sabat
Organometallics 2013 Volume 32(Issue 6) pp:1797-1806
Publication Date(Web):February 27, 2013
DOI:10.1021/om301219t
The Fe(II) complex Cp*Fe(CO)(NCMe)Ph (Cp* = η5-pentamethylcyclopentadienyl) is shown to mediate facile and highly regioselective C–H activation of aromatic substrates including benzene, furan, thiophene, thiazole, and 2-methylfuran. Experimental and computational evidence suggest a mechanism for C–H activation that involves NCMe dissociation, multiple spin intersystem crossings, C–H bond coordination, and C–H bond cleavage by a σ-bond metathesis reaction.
Co-reporter:Thomas Ghebreghiorgis, Brian H. Kirk, Aaron Aponick, and Daniel H. Ess
The Journal of Organic Chemistry 2013 Volume 78(Issue 15) pp:7664-7673
Publication Date(Web):July 17, 2013
DOI:10.1021/jo4012283
Density functional calculations and experiments were used to examine mechanisms of Pd(II) catalyzed intramolecular cyclization and dehydration in acyclic and bicyclic monoallylic diols, a formal SN2′ reaction. In contrast to the previously proposed syn-oxypalladation mechanism for acyclic monoallylic diols, calculations and experiments strongly suggest that hydrogen bonding templates a hydroxyl group and Pd addition across the alkene and provides a low energy pathway via anti-addition (anti-oxypalladation) followed by intramolecular proton transfer and anti-elimination of water. This anti-addition, anti-elimination pathway also provides a simple rationale for the observed stereospecificity. For bicyclic monoallylic diol compounds, Pd(II) is capable of promoting either anti- or syn-addition. In addition, palladium chloride ligands can mediate proton transfer to promote dehydration when direct intramolecular proton transfer between diol groups is impossible.
Co-reporter:Thomas Ghebreghiorgis ; Berenger Biannic ; Brian H. Kirk ; Daniel H. Ess ;Aaron Aponick
Journal of the American Chemical Society 2012 Volume 134(Issue 39) pp:16307-16318
Publication Date(Web):September 4, 2012
DOI:10.1021/ja306333a
Density functional calculations and experiment were used to examine the mechanism, reactivity, and origin of chirality transfer in monophosphine Au-catalyzed monoallylic diol cyclization reactions. The lowest energy pathway for cyclization involves a two-step sequence that begins with intramolecular C–O bond formation by anti-addition of the non-allylic hydroxyl group to the Au-coordinated alkene followed by concerted hydrogen transfer/anti-elimination to liberate water. Concerted SN2′-type transition states were found to be significantly higher in energy. The two-step cyclization pathway is extremely facile due to hydrogen bonding between diol groups that induces nucleophilic attack on the alkene and then proton transfer between diol groups after C–O bond formation. Importantly, intramolecular proton transfer and elimination provides an extremely efficient avenue for catalyst regeneration from the Au–C σ-bond intermediate, in contrast to other Au-catalyzed cyclization reactions where this intermediate severely restricts catalyst turnover. The origin of chirality transfer and the ensuing alkene stereochemistry is also the result of strong hydrogen-bonding interactions between diol groups. In the C–O bond-forming step, requisite hydrogen bonding biases the tethered nucleophilic moiety to adopt a chair-like conformation with substituents in either axial or equatorial positions, dictating the stereochemical outcome of the reaction. Since this hydrogen bonding is maintained throughout the course of the reaction, establishment of the resultant olefin geometry is also attributed to this templating effect. These computational conclusions are supported by experimental evidence employing bicyclic systems to probe the facial selectivity.
Co-reporter:Thomas C. Cook, Merritt B. Andrus, and Daniel H. Ess
Organic Letters 2012 Volume 14(Issue 23) pp:5836-5839
Publication Date(Web):November 19, 2012
DOI:10.1021/ol3026582
Density functional theory was used to model glycinate enolate binding and enantiomeric allylation transition states mediated by the cinchonidinium phase-transfer catalyst 2. Transition states show oxy-anion-ammonium interactions in contrast to π-face interactions in the ground states. The details of stereoselectivity are described within the quaternary ammonium-tetrahedron face model.
Co-reporter:Alban Petit, Josh Flygare, Alex T. Miller, Gerrit Winkel, and Daniel H. Ess
Organic Letters 2012 Volume 14(Issue 14) pp:3680-3683
Publication Date(Web):July 10, 2012
DOI:10.1021/ol301521n
Density functional calculations reveal that the stability of developing metal aryl bonds in Pd(II)-acetate C–H activation transition states determines regioselectivity in arene and heteroarene compounds. This kinetic–thermodynamic connection explains the general preference for activation of the strongest C–H bond and provides the possibility for regioselectivity prediction.
Co-reporter:Deepa Devarajan, T. Brent Gunnoe, and Daniel H. Ess
Inorganic Chemistry 2012 Volume 51(Issue 12) pp:6710-6718
Publication Date(Web):June 4, 2012
DOI:10.1021/ic300350k
Density functional and correlated ab initio methods were used to calculate, compare, and analyze bonding interactions in late-transition-metal alkyl and heteroatom complexes (M–X). The complexes studied include: (DMPE)Pt(CH3)(X) (DMPE = 1,2-bis(dimethylphosphino)ethane), Cp*Ru(PMe3)2(X) (Cp* = pentamethylcyclopentadienyl), (DMPE)2Ru(H)(X), (Tp)(CO)Ru(Py)(X) (Tp = trispyrazolylborate), (PMe3)2Rh(C2H4)(X), and cis-(acac)2Ir(Py)(X) (acac = acetylacetonate). Seventeen X ligands were analyzed that include alkyl (CR3), amido (NR2), alkoxo (OR), and fluoride. Energy decomposition analysis of these M–X bonds revealed that orbital charge transfer stabilization provides a straightforward model for trends in bonding along the alkyl to heteroatom ligand series (X = CH3, NH2, OH, F). Pauli repulsion (exchange repulsion), which includes contributions from closed-shell dπ-pπ repulsion, generally decreases along the alkyl to heteroatom ligand series but depends on the exact M–X complexes. It was also revealed that stabilizing electrostatic interactions generally decrease along this ligand series. Correlation between M–X and H–X bond dissociation energies is good with R2 values between 0.7 and 0.9. This correlation exists because for both M–X and H–X bonds the orbital stabilization energies are a function of the orbital electronegativity of the X group. The greater than 1 slope when correlating M–X and H–X bond dissociation energies was traced back to differences in Pauli repulsion and electrostatic stabilization.
Co-reporter:Deepa Devarajan and Daniel H. Ess
Inorganic Chemistry 2012 Volume 51(Issue 11) pp:6367-6375
Publication Date(Web):May 16, 2012
DOI:10.1021/ic3006426
Density functional theory and absolutely localized molecular orbital energy decomposition analysis calculations were used to calculate and analyze dihydrogen activation transition states and reaction pathways. Analysis of a variety of transition-metal complexes with d0, d6, d8, and d10 orbital occupation with a diverse range of metal ligands reveals that for transition states, akin to dihydrogen σ complexes, there is a continuum of activated H–H bond lengths that can be classified as “dihydrogen” (0.8–1.0 Å), “stretched or elongated” (1.0–1.2 Å), and “compressed dihydride” (1.2–1.6 Å). These calculations also quantitatively for the first time reveal that the extent to which H2 is activated in the transition-structure geometry depends on back-bonding orbital interactions and not forward-bonding orbital interactions. This is true regardless of the mechanism or whether the metal ligand complex acts as an electrophile, ambiphile, or nucleophile toward dihydrogen.
Co-reporter:Daniel H. Ess
Journal of Chemical Education 2012 Volume 89(Issue 6) pp:817-818
Publication Date(Web):April 5, 2012
DOI:10.1021/ed2005856
Modern organic chemistry textbooks contain qualitative Lewis-structure drawings of transition structures. Missing from these drawings are estimates of partial bond lengths. A catalog of accurate density functional ground and transition structures for radical abstraction, substitution, elimination, oxidation, and pericyclic reactions is provided in the Supporting Information and discussed here. These xyz transition-structure coordinates can be conveniently and easily viewed in a variety of free and commercial viewers and provide the ability to quantitatively discuss bonding changes in organic reactions. In addition to the lowest-energy-pathway transition structures, this catalog also provides higher-energy-pathway transition structures, for example, backside and frontside SN2 transition structures as well as anti and syn E2 transition structures.Keywords: Computational Chemistry; Computer-Based Learning; Molecular Modeling; Organic Chemistry; Second-Year Undergraduate;
Co-reporter:Daniel H. Ess and Thomas C. Cook
The Journal of Physical Chemistry A 2012 Volume 116(Issue 20) pp:4922-4929
Publication Date(Web):May 11, 2012
DOI:10.1021/jp300633j
Here we present and test several computational prescriptions for calculating singlet–triplet (ST) gap energies and bond dissociation curves for open-shell singlet diradicals using economical unrestricted single reference type calculations. For ST gap energies from Slipchenko and Krylov’s atom and molecule test set (C, O, Si, NH, NF, OH+, O2, CH2, and NH2+) spin unrestricted Hartree–Fock and MP2 energies result in errors greater than 15 kcal/mol. However, spin–projected (SP) Hartree–Fock theory in combination with spin-component-scaled (SCS) or scaled-opposite-spin (SOS) second-order perturbation theory gives ST gap energies with a mean unsigned error (MUE) of less than 2 kcal/mol. Density functionals generally give poor results for unrestricted energies and only the ωB97X-D, the M06, and the M06-2X functionals provide reasonable accuracy after spin-projection with MUE values of 4.7, 4.3, and 3.0 kcal/mol, respectively, with the 6-311++G(2d,2p) basis set. We also present a new one parameter hybrid density functional, diradical-1 (DR-1), based on Adamo and Barone’s modified PW exchange functional with the PW91 correlation functional. This DR-1 method gives a mean error (ME) of 0.0 kcal/mol and a MUE value of 1.3 kcal/mol for ST gap energies. As another test of unrestricted methods the bond dissociation curves for methane (CH4) and hydrofluoric acid (H–F) were calculated with the M06-2X, DR-1, and ωB97X-D density functionals. All three of these functionals give reasonable results for the methane C–H bond but result in errors greater than 50 kcal/mol for the H–F bond dissociation. Spin-projection is found to significantly degrade bond dissociation curves past ∼2.2 Å. Although unrestricted Hartree–Fock theory provides a very poor description of H–F bond dissociation, unrestricted SCS-MP2 and SOS-MP2 methods give accurate results.
Co-reporter:Boris Vabre, Melinda L. Lambert, Alban Petit, Daniel H. Ess, and Davit Zargarian
Organometallics 2012 Volume 31(Issue 17) pp:6041-6053
Publication Date(Web):August 23, 2012
DOI:10.1021/om3003784
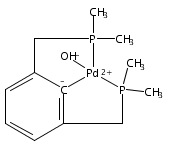
This report describes the results of a combined experimental and computational investigation on the kinetics and mechanism of the C–H metalation step involved in the formation of PCP- and POCOP-type complexes of nickel. The kinetics of the C–H nickelation reaction was probed through competition studies involving two ligands reacting with a substoicheometric quantity of {(i-PrCN)NiBr2}n. These experiments have confirmed that metalation is more facile for aromatic ligands 1,3-(i-Pr2PE)2C6H4 vs their aliphatic counterparts 1,3-(i-Pr2PECH2)2CH2 (sp2 C–H > sp3 C–H; E = O, CH2), ligands bearing phosphine moieties vs those with phosphinite moieties (PCP > POCOP), ligands bearing P substituents i-Pr2P vs t-Bu2P and Ph2P, and POCsp2OP ligands 1,3-(i-Pr2PO)2C6RnH4–n bearing electron-donating vs electron-withdrawing substituents (p-OMe ≈ m-OMe > p-Me > m-CO2Me > p-CO2Me > m,m-Cl2). Among the latter, there is a 6-fold difference in C–H metalation rate between ligands bearing p-OMe and p-COOMe, whereas the most readily metalating ligand, 1,3-(i-Pr2PCH2)2C6H4, is metalated ca. 270 times more readily relative to the least reactive ligand, 1,3-(i-Pr2POCH2)2CH2. Density functional calculations indicate that PCP- or POCOP-type pincer ligands react with NiBr2 to generate nonmetalated intermediates that form the corresponding pincer complexes via a two-step mechanism involving an ionic dissociation of the bromide to give a tight ion pair intermediate, followed by bromide-assisted deprotonation of the C–H bond. The type of structure adopted by the nonmetalated intermediates (mono- or dinuclear; tetrahedral, cis or trans square planar) and the energy barriers for the metalation transition states depend on the steric properties of the PR2 moiety. The presence of a base that can neutralize the HBr generated in the metalation step is crucial for rendering the metalation process exergonic. One rationale for the more facile metalation of PCP ligands in comparison to their POCOP counterparts is the greater donor character of phosphine moieties, which allows a more effective stabilization of the coordination and metalation transition states wherein the strongly donor halide ligand is displaced by a much weaker C–H bond donor. The aromatic ligands metalate more readily than their aliphatic analogues for multiple reasons, including the higher ground state energy of the nonmetalated intermediates formed with aromatic ligands, the stronger Csp2–Ni bond formed via metalation, and the more stabilized anionic charge on the C atom being metalated.
Co-reporter:Preston S. Stewart;Linda Rodriguez
Journal of Physical Organic Chemistry 2011 Volume 24( Issue 12) pp:1222-1228
Publication Date(Web):
DOI:10.1002/poc.1850
Abstract
Isomerization energies for hexenes (C6H12) were evaluated with ab initio (Hartree–Fock (HF), MP2, SCS-MP2, and CCSD(T)) and several density functional approximation (DFA) methods. CCSD(T)/6-311+G(2d,p) energies were taken as a benchmark standard. The HF method incorrectly predicts that monosubstituted alkenes are more stable than multiply-substituted alkenes. DFAs generally predict the correct stability trends of alkenes (mono-, < di-, < tri-, < tetra-substituted alkenes) but errors in popular functionals, such as B3LYP, can be as large as errors found for alkane hydrocarbon thermochemistries. Some of the HF error is traced back to deficiencies in modeling 1,3-geminal and 1,4-vicinal alkyl–alkyl group interactions, called vinylbranches, and changes in CC and CH bond types (sp3–sp2 CC to sp3–sp3 CC and sp3 CH to sp2 CH). The latter is shown to be more significant. Comparison of CCSD(T) energies of trans-2-butene with 2-methylpropylene and cis-2-butene suggests that geminal vinylbranches are stabilizing while vicinal vinylbranches are destabilizing. B3LYP and other DFAs have much smaller errors than HF theory due to inclusion of correlation energy that better reproduces bond type changes. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:A. George Johnson, Brad M. Loertscher, Adam R. Moeck, Sam S. Matthews, Daniel H. Ess, Steven L. Castle
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 9) pp:2706-2710
Publication Date(Web):1 May 2011
DOI:10.1016/j.bmcl.2010.11.121
The scope of enantioselective allylations employing Nakamura’s allylzinc–bisoxazoline reagent was examined by performing allylations of a selection of readily available ketones. Low-to-moderate ee’s were observed, and a computational study was conducted to rationalize the results. Examination of transition structures of previously performed allylations that proceeded with high ee revealed the importance of both local and global control elements in these successful reactions. The ability of density functional theory methods to estimate the enantioselectivity of these asymmetric ketone allylations was established. All allylations that were studied computationally exhibited low (<5 kcal/mol) activation barriers, a result that is consistent with the highly reactive nature of Nakamura’s reagent.An investigation of the scope of enantioselective ketone allylations employing Nakamura’s chiral allylzinc–bisoxazoline reagent is described, along with the development of a theoretical model to explain the results.
Co-reporter:Brian H. Kirk, Daniel H. Ess
Tetrahedron Letters 2011 Volume 52(Issue 12) pp:1245-1249
Publication Date(Web):23 March 2011
DOI:10.1016/j.tetlet.2011.01.026
The Diels–Alder reaction of substituted cyclohexadienes with substituted phenylacetylenes offers an attractive alternative for the synthesis of biaryl compounds via a two-step cycloaddition/cycloelimination pathway. Quantum mechanical calculations using B3LYP and M06-2X density functional methods for the reaction of 2-chloro-6-nitrophenylacetylene with 1-carbomethoxy-cyclohexadiene show the reaction proceeds by a stepwise diradical [4+2] cycloaddition followed by concerted [2+4] cycloelimination of ethylene. [2+2] cycloadducts are also the result of stepwise addition. [2+2] cycloadducts isomerize to [4+2] cycloadducts via diradical pathways, which involve the same diradical intermediate in cycloaddition. There is also a competitive conrotatory ring opening followed by trans-cis double bond isomerization pathway of the [4.2.0] bicycle (the [2+2] cycloadduct) to give the cis,cis,cis-1,3,5-cyclooctatriene.
Co-reporter:Daniel H. Ess, Erin R. Johnson, Xiangqian Hu, and Weitao Yang
The Journal of Physical Chemistry A 2011 Volume 115(Issue 1) pp:76-83
Publication Date(Web):December 9, 2010
DOI:10.1021/jp109280y
Open-shell singlet diradicals are difficult to model accurately within conventional Kohn−Sham (KS) density-functional theory (DFT). These methods are hampered by spin contamination because the KS determinant wave function is neither a pure spin state nor an eigenfunction of the S2 operator. Here we present a theoretical foray for using single-reference closed-shell ground states to describe diradicals by fractional-spin DFT (FS-DFT). This approach allows direct, self-consistent calculation of electronic properties using the electron density corresponding to the proper spin eigenfunction. The resulting FS-DFT approach is benchmarked against diradical singlet−triplet gaps for atoms and small molecules. We have also applied FS-DFT to the singlet−triplet gaps of hydrocarbon polyacenes.
Co-reporter:Corey S. Ellis and Daniel H. Ess
The Journal of Organic Chemistry 2011 Volume 76(Issue 17) pp:7180-7185
Publication Date(Web):August 4, 2011
DOI:10.1021/jo201234f
The key platinum mediated C–H bond activation and functionalization steps in the synthesis of (−)-rhazinilam (Johnson, J. A.; Li, N.; Sames, D. J. Am. Chem. Soc.2002, 124, 6900) were investigated using the M06 and B3LYP density functional approximation methods. This computational study reveals that ethyl group dehydrogenation begins with activation of a primary C–H bond in preference to a secondary C–H bond in an insertion/methane elimination pathway. The C–H activation step is found to be reversible while the methane elimination (reductive elimination) transition state controls rate and diastereoselectivity. The chiral oxazolinyl ligand induces ethyl group selectivity through stabilizing weak interactions between its phenyl group (or cyclohexyl group) and the carboxylate group. After C–H activation and methane elimination steps, Pt–C bond functionalization occurs through β-hydride elimination to give the alkene platinum hydride complex.
Co-reporter:Daniel H. Ess, Shubin Liu, and Frank De Proft
The Journal of Physical Chemistry A 2010 Volume 114(Issue 49) pp:12952-12957
Publication Date(Web):November 18, 2010
DOI:10.1021/jp108577g
Branched alkane hydrocarbons are thermodynamically more stable than straight-chain linear alkanes. This thermodynamic stability is also manifest in alkane bond separation energies. To understand the physical differences between branched and linear alkanes, we have utilized a novel density functional theory (DFT) definition of steric energy based on the Weizäcker kinetic energy. Using the M06-2X functional, the total DFT energy was partitioned into a steric energy term (Es[ρ]), an electrostatic energy term (Ee[ρ]), and a fermionic quantum energy term (Eq[ρ]). This analysis revealed that branched alkanes have less (destabilizing) DFT steric energy than linear alkanes. The lower steric energy of branched alkanes is mitigated by an equal and opposite quantum energy term that contains the Pauli component of the kinetic energy and exchange-correlation energy. Because the steric and quantum energy terms cancel, this leaves the electrostatic energy term that favors alkane branching. Electrostatic effects, combined with correlation energy, explains why branched alkanes are more stable than linear alkanes.
Co-reporter:Daniel H. Ess, T. Brent Gunnoe, Thomas R. Cundari, William A. Goddard III, and Roy A. Periana
Organometallics 2010 Volume 29(Issue 24) pp:6801-6815
Publication Date(Web):December 3, 2010
DOI:10.1021/om100974q
Mid to late transition metal complexes that break hydrocarbon C−H bonds by transferring the hydrogen to a heteroatom ligand while forming a metal−alkyl bond offer a promising strategy for C−H activation. Here we report a density functional (B3LYP, M06, and X3LYP) analysis of cis-(acac)2MX and TpM(L)X (M = Ir, Ru, Os, and Rh; acac = acetylacetonate, Tp = tris(pyrazolyl)borate; X = CH3, OH, OMe, NH2, and NMe2) systems for methane C−H bond activation reaction kinetics and thermodynamics. We address the importance of whether a ligand lone pair provides an intrinsic kinetic advantage through possible electronic dπ−pπ repulsions for M−OR and M−NR2 systems versus M−CH3 systems. This involves understanding the energetic impact of the X ligand group on ligand loss, C−H bond coordination, and C−H bond cleavage steps as well as understanding how the nucleophilicity of the ligand X group, the electrophilicity of the transition metal center, and cis-ligand stabilization effect influence each of these steps. We also explore how spectator ligands and second- versus third-row transition metal centers impact the energetics of each of these C−H activation steps.
Co-reporter:Daniel H. Ess, William A. Goddard III, and Roy A. Periana
Organometallics 2010 Volume 29(Issue 23) pp:6459-6472
Publication Date(Web):October 29, 2010
DOI:10.1021/om100879y
The potential energy and interaction energy profiles for metal- and metal−ligand-mediated alkane C−H bond activation were explored using B3LYP density functional theory (DFT) and the absolutely localized molecular orbital energy decomposition analysis (ALMO-EDA). The set of complexes explored range from late transition metal group 10 (Pt and Pd) and group 11 (Au) metal centers to group 7−9 (Ir, Rh, Ru, and W) metal centers as well as a group 3 Sc complex. The coordination geometries, electron metal count (d8, d6, d4, and d0), and ligands (N-heterocycles, O-donor, phosphine, and Cp*) are also diverse. Quantitative analysis using ALMO-EDA of both directions of charge-transfer stabilization (occupied to unoccupied orbital stabilization) energies between the metal−ligand fragment and the coordinated C−H bond in the transition state for cleavage of the C−H bond allows classification of C−H activation reactions as electrophilic, ambiphilic, or nucleophilic on the basis of the net direction of charge-transfer energy stabilization. This bonding pattern transcends any specific mechanistic or bonding paradigm, such as oxidative addition, σ-bond metathesis, or substitution. Late transition metals such as Au(III), Pt(II), Pd(II), and Rh(III) metal centers with N-heterocycle, halide, or O-donor ligands show electrophilically dominated reaction profiles with forward charge-transfer from the C−H bond to the metal, leading to more stabilization than reverse charge transfer from the metal to the C−H bond. Transition states and reaction profiles for d6 Ru(II) and Ir(III) metals with Tp and acac ligands were found to have nearly equal forward and reverse charge-transfer energy stabilization. This ambiphilic region also includes the classically labeled electrophilic cationic species Cp*(PMe3)Ir(Me). Nucleophilic character, where the metal to C−H bond charge-transfer interaction is most stabilizing, was found in metathesis reactions with W(II) and Sc(III) metal center complexes in reactions as well as late transition metal Ir(I) and Rh(I) pincer complexes that undergo C−H bond insertion. Comparison of pincer ligands shows that the PCP ligand imparts more nucleophilic character to an Ir metal center than a deprotonated PNP ligand. The PCP and POCOP ligands do not show a substantial difference in the electronics of C−H activation. It was also found that Rh(I) is substantially more nucleophilic than Ir(I). Lastly, as a qualitative approximation, investigation of transition-state fragment orbital energies showed that relative frontier orbital energy gaps correctly reflect electrophilic, ambiphilic, or nucleophilic charge-transfer stabilization patterns.
Co-reporter:Ryan W. Carlsen and Daniel H. Ess
Dalton Transactions 2016 - vol. 45(Issue 24) pp:NaN9840-9840
Publication Date(Web):2016/02/19
DOI:10.1039/C6DT00256K
Transition metal heterobimetallic complexes with dative metal–metal interactions have the potential for novel fast reactivity. There are few studies that both compare the reactivity of different metal centers in heterobimetallic complexes and compare bimetallic reactivity to monometallic reactivity. Here we report density-functional calculations that show the reactivity of [Cl2Ti(NtBuPPh2)2MII(η3-methallyl)] heterobimetallic complexes for allylic amination follows M = Ni > Pd > Pt. This reactivity trend was not anticipated since the amine addition transition state involves MII to M0 reduction and this could disadvantage Ni. Comparison of heterobimetallic complexes to the corresponding monometallic (CH2)2(NtBuPPh2)2MII(η3-methallyl) complexes reveals that this reactivity trend is due to the bimetallic interaction and that the bimetallic interaction significantly lowers the barrier height for amine addition by >10 kcal mol−1. The impact of the early transition metal center on the amination addition barrier height depends on the late transition metal center. The lowest barrier heights for this reaction occur when late and early transition metal centers are from the same periodic table row.
.jpg)

![2,3,5-TRIAZABICYCLO[2.2.2]OCTA-2,5-DIENE](http://img.cochemist.com/ccimg/871300/871260-70-7.png)
![2,3,5-TRIAZABICYCLO[2.2.2]OCTA-2,5-DIENE](http://img.cochemist.com/ccimg/871300/871260-70-7_b.png)
![Propylidene, 1-[bis(1-methylethyl)amino]-2,2-dimethyl-](http://img.cochemist.com/ccimg/737800/737756-00-2.png)
![Propylidene, 1-[bis(1-methylethyl)amino]-2,2-dimethyl-](http://img.cochemist.com/ccimg/737800/737756-00-2_b.png)







![CARBAMIC ACID, [(METHYLSULFONYL)OXY]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/91000/90966-54-4.png)
![CARBAMIC ACID, [(METHYLSULFONYL)OXY]-, PHENYLMETHYL ESTER](http://img.cochemist.com/ccimg/91000/90966-54-4_b.png)







![2-Azabicyclo[2.2.2]octa-2,5-diene](http://img.cochemist.com/ccimg/57200/57163-98-1.png)
![2-Azabicyclo[2.2.2]octa-2,5-diene](http://img.cochemist.com/ccimg/57200/57163-98-1_b.png)





![2,3-Diazabicyclo[2.2.2]octa-2,5-diene](http://img.cochemist.com/ccimg/37500/37436-20-7.png)
![2,3-Diazabicyclo[2.2.2]octa-2,5-diene](http://img.cochemist.com/ccimg/37500/37436-20-7_b.png)









![bicyclo[2.2.2]octa-2,5-diene](http://img.cochemist.com/ccimg/600/500-23-2.png)
![bicyclo[2.2.2]octa-2,5-diene](http://img.cochemist.com/ccimg/600/500-23-2_b.png)