Co-reporter:Qingxi Meng;Fen Wang
Journal of Molecular Modeling 2017 Volume 23( Issue 1) pp:11
Publication Date(Web):21 December 2016
DOI:10.1007/s00894-016-3186-7
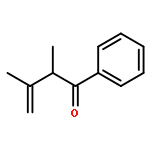
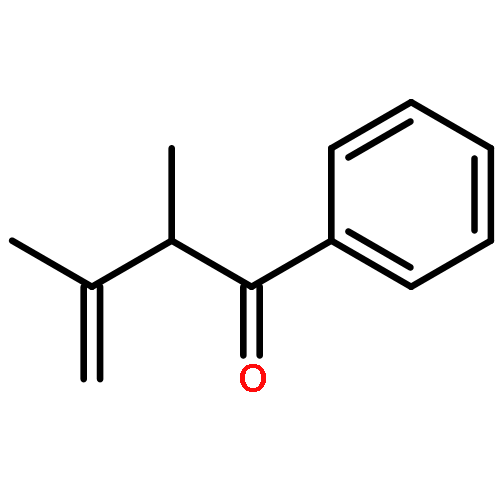
Density functional calculations have been applied to study and elucidate nickel(0)/N-heterocyclic carbene-catalyzed intramolecular alkene hydroacylation. The calculations showed that nickel(0)-catalyzed intramolecular alkene hydroacylation involved four potential reaction channels (I, II, III, and IV), and pathway IV was predicted to be more favorable than the other three. Two pathways, I and II, had three steps (oxidative addition, hydrogen migration, reductive elimination), and the rate-determining step was hydrogen migration. Pathway III proceeded through oxidative cyclization, β-hydride elimination, and hydrogen migration, and the rate-determining step was β-hydride elimination. Pathway IV included four steps (oxidative cyclization, dimerization, β-hydride elimination, hydrogen migration), and the rate-determining step was again β-hydride elimination. Oxidative cyclization was easy and led to rapid dimerization, greatly reducing the free energy of β-hydride elimination. The binuclear nickelacycle intermediate was observed in Ogoshi’s experiments, and it was identified by nuclear magnetic resonance (NMR). The dominant product was the five-membered benzocyclic ketone p1. All results agreed with Ogoshi’s experiments.
Co-reporter:Qingxi Meng;Fen Wang
Journal of Molecular Modeling 2016 Volume 22( Issue 3) pp:
Publication Date(Web):2016 March
DOI:10.1007/s00894-016-2930-3
Density functional theory (DFT) was used to study the cobalt(I)-catalyzed enantioselective intramolecular hydroacylation of ketones and alkenes. All intermediates and transition states were fully optimized at the M06/6-31G(d,p) level (LANL2DZ(f) for Co). The results demonstrated that the ketone and alkene present different reactivities in the enantioselective hydroacylation. In ketone hydroacylation catalyzed by the cobalt(I)–(R,R)-Ph-BPE complex, reaction channel “a” to (R)-phthalide was more favorable than channel “b” to (S)-phthalide. Hydrogen migration was both the rate-determining and chirality-limiting step, and this step was endothermic. In alkene hydroacylation catalyzed by the cobalt(I)–(R,R)-BDPP complex, reaction channel “c” leading to the formation of (S)-indanone was the most favorable, both thermodynamically and kinetically. Reductive elimination was the rate-determining step, but the chirality-limiting step was hydrogen migration, which occurred easily. The results also indicated that the alkene hydroacylation leading to (S)-indanone formation was more energetically favorable than the ketone hydroacylation that gave (R)-phthalide, both thermodynamically and kinetically.
Co-reporter:Qingxi Meng;Fen Wang;Hongzong Yin
Journal of Physical Organic Chemistry 2015 Volume 28( Issue 6) pp:431-436
Publication Date(Web):
DOI:10.1002/poc.3432
Density functional theory (DFT) was used to investigate computationally cobalt(I)-catalyzed hydroacylation of vinylsilanes and alkyl aldehydes to give ketones. Calculation indicated that cobalt(I)-catalyzed hydroacylation had eight possible reaction pathways. In the cobalt-hydride complexes IM2a and IM2b, the hydrogen migration occurred prior to the carbon–carbon bond-forming reaction. In the complexes IM3a1 and IM3b1, the carbonyl elimination reaction occurred prior to the direct reductive elimination reaction. In the cobalt–carbonyl complexes IM4a and IM4b, the carbonyl insertion reaction was much easier to achieve than the decarbonylation reaction. The dominant reaction pathway was the reaction channel IM1a TS1a IM2a TS2a1 IM3a1 TS4a IM4a TS5a IM5a TS6a IM6a, and the reductive elimination reaction was the rate-determining step for this channel, so the dominant product predicted theoretically was the linear ketone. Furthermore, the solvation effect was remarkable, and it decreased generally the free energies of the species. Copyright © 2015 John Wiley & Sons, Ltd.
Co-reporter:Fen Wang;Shuhua Zhu;Hongzong Yin
Journal of Molecular Modeling 2015 Volume 21( Issue 8) pp:
Publication Date(Web):2015 August
DOI:10.1007/s00894-015-2754-6
Density functional theory (DFT) was used to investigate nickel-catalyzed ring-opening hydroacylation of methylenecyclopropanes and benzaldehydes. The results indicated that the Ni-P(n-Bu)3 complex exhibited much more excellent catalysis than the other two complexes (Ni-PMe3 and Ni-P(t-Bu)3). The hydrogen migration was the rate-determining step, and the β-carbon elimination was the chirality-limiting step. The dominant product was a (S,S)- cis ketone. The phosphine ligand P(n-Bu)3 changed the rate-determining step, and greatly decreased the free energies of the rate-determining step and chirality-limiting step. The use of P(n-Bu)3 generally decreased the free energies of the intermediates and transition states. The possible role of P(n-Bu)3 was the transformation of the electron and geometry structures of those intermediates and transition states.
Co-reporter:Fen Wang, Qingxi Meng, Ming Li
Journal of Organometallic Chemistry 2014 Volume 753() pp:1-8
Publication Date(Web):1 March 2014
DOI:10.1016/j.jorganchem.2013.12.012
•We studied the different reactivity of two π bonds of isoprene.•We studied the role of the ligands hydride, carbonyl, chloride, and triphenylphosphine of RuHCl(CO)(PPh3)3.•We studied the effect of the di-ruthenium complexes.Density functional theory (DFT) was used to investigate ruthenium hydride-catalyzed addition reactions of benzaldehydes to isoprenes. Calculation indicated that two π bonds of isoprene had different reactivity, and the less substituted one had better reactivity. The role of the ligands hydride, carbonyl, chloride, and triphenylphosphine of RuHCl(CO)(PPh3)3 were studied in our present work. The hydride was active, and promoted this reaction. The carbonyl or chloride was not used as a carrier of hydrogen migration reaction, so they could not change the reaction channels. The only role of them was the transformation of the electron and geometry structures of those intermediates and transition states. The triphenylphosphine decreased generally the free energies of the intermediates and transition states. The di-ruthenium complexes were not an efficient catalyst.Calculation indicated that two π bonds of isoprene had different reactivity, and the less substituted one had better reactivity. The role of the ligands hydride, carbonyl, chloride, and triphenylphosphine of RuHCl(CO)(PPh3)3 were studied in our present work.
Co-reporter:Qingxi Meng;Fen Wang;Ming Li
Journal of Molecular Modeling 2013 Volume 19( Issue 6) pp:2225-2234
Publication Date(Web):2013 June
DOI:10.1007/s00894-013-1772-5
Density functional theory (DFT) was used to investigate cobalt(0)-catalyzed intramolecular hydroacylation of 4-pentenal. The calculated results indicated the involvement of five possible reaction pathways: the formation of cyclopentanone, cyclobutanone, butylenes, cyclobutane, and cyclopropane, respectively. The former two are pathways of Co(0)-catalyzed intramolecular hydroacylation, while the latter three are pathways of decarbonylation. The formation of cyclopentanone was the most favorable channel, and the oxidative addition reaction of 4-pentenal was the rate-determining step. Hence, the dominant product predicted theoretically was cyclopentanone, which was consistent with experimental results. Solvation had a significant effect, and greatly decreased the free energies of all intermediates and transition states.
Co-reporter:Qingxi Meng;Fen Wang;Ming Li
Journal of Molecular Modeling 2012 Volume 18( Issue 12) pp:4955-4963
Publication Date(Web):2012 December
DOI:10.1007/s00894-012-1493-1
Density functional theory (DFT) was used to investigate the ruthenium hydride-catalyzed regioselective addition reactions of benzaldehyde to isoprene leading to the branched β,γ-unsaturated ketone. All intermediates and the transition states were optimized completely at the B3LYP/6-31 G(d,p) level (LANL2DZ(f) for Ru, LANL2DZ(d) for P and Cl). Calculated results indicated that three catalysts RuHCl(CO)(PMe3)3 (1), RuH2(CO)(PMe3)3 (2), and RuHCl(PMe3)3 (3) exhibited different catalysis, and the first was the most excellent. The most favorable reaction pathway included the coordination of 1 to the less substituted olefin of isoprene, a hydrogen transfer reaction from ruthenium to the carbon atom C1, the complexation of benzaldehyde to ruthenium, the carbonyl addition, and the hydride elimination reaction. The carbonyl addition was the rate-determining step. The dominant product was the branched β,γ-unsaturated ketone. Furthermore, the presence of one toluene molecule lowered the activation free energy of the transition state of the carbonyl addition by hydrogen bonds between the protons of toluene and the chlorine, carbonyl oxygen of the ruthenium complex. On the whole, the solvent effect decreased the free energies of the species.
Co-reporter:Fen Wang;Jianmin Wang;Ming Li
Structural Chemistry 2009 Volume 20( Issue 1) pp:129-137
Publication Date(Web):2009 February
DOI:10.1007/s11224-008-9352-5
The alkynylation of nitrones catalyzed by chiral zinc(II)-complexes was studied by means of the density functional theory (DFT). All the intermediates and transition states were optimized completely at B3LYP/6-31G(d,p) level. Calculation results confirm that this alkynylation of nitrones was endothermic and the total absorbed energy was about 14 kJ/mol. The formation of the M5 complexes was the rate-determining step and the chirality-limiting step for this alkynylation. The transition states TS3-re involve a H(8)–O(2)–Zn(6)–C(9)–C(14)–N(13)–O(12) 7-membered ring, and thus the transition states TS3-si involve a Zn(6)–C(9)–C(14)–N(13)–O(12) 5-membered ring. The dominant reaction is the attack of nitrones from the re-surface of M4. The reactivity of anti-nitrones was stronger than that of syn-nitrones for zinc-catalyzed alkynylation of nitrones. The dominant reaction channel was cata → TS1b → M1b → M2 → M3b → TS2b → M4b → TS3b1-anti-re → M5b1-anti-re → Pro-R. The dominant products predicted theoretically were of R-chirality, (2R)-N-hydroxy-N-methylpent-3-yn-2-amine.
Co-reporter:Qingxi Meng, Fen Wang, Xiangjin Qu, Jie Zhou, Ming Li
Journal of Molecular Structure: THEOCHEM 2007 Volume 815(1–3) pp:157-163
Publication Date(Web):1 August 2007
DOI:10.1016/j.theochem.2007.03.027
Density functional theory has been used to study the geometries, properties and reactivity of the iron(II)–carbene complexes Cp(CO)(L)FeCHR, L = CO, PMe3, R = Me, OMe, ph, CO2Me. Calculation results confirm that the iron–carbene complexes have two types of geometries: the Fe–C(carbene) bonds of the iron–carbene complexes A, D, E, and H (R = Me, CO2Me) show a strong double-bonded character, and thus the Fe–C(carbene) bonds of the complexes B, C, F, and G (R = OMe, ph) show a strong single-bonded character. The Fe–C(carbene) bonds, and the iron and carbon atoms of Fe–C(carbene) bonds are the reactive center for the iron–carbene complexes. The geometry, electronic structure, and frontier orbitals (HOMO and LUMO) are discussed in detail.

![(2r,5r)-1-[2-[(2r,5r)-2,5-diphenylphospholan-1-yl]ethyl]-2,5-diphenylphospholane](http://img.cochemist.com/ccimg/528600/528565-79-9.png)
![(2r,5r)-1-[2-[(2r,5r)-2,5-diphenylphospholan-1-yl]ethyl]-2,5-diphenylphospholane](http://img.cochemist.com/ccimg/528600/528565-79-9_b.png)


![Phosphine,1,1'-[(1R,3R)-1,3-dimethyl-1,3-propanediyl]bis[1,1-diphenyl-](http://img.cochemist.com/ccimg/96200/96183-46-9.png)
![Phosphine,1,1'-[(1R,3R)-1,3-dimethyl-1,3-propanediyl]bis[1,1-diphenyl-](http://img.cochemist.com/ccimg/96200/96183-46-9_b.png)