Co-reporter:Weijie You;Dr. Dante Rotili;Dr. Tie-Mei Li;Dr. Christian Kambach;Dr. Marat Meleshin; Dr. Mike Schutkowski; Dr. Katrin F. Chua; Dr. Antonello Mai; Dr. Clemens Steegborn
Angewandte Chemie International Edition 2017 Volume 56(Issue 4) pp:1007-1011
Publication Date(Web):2017/01/19
DOI:10.1002/anie.201610082
AbstractSirtuins are protein deacylases regulating metabolism and stress responses, and are implicated in aging-related diseases. Small molecule activators for the human sirtuins Sirt1-7 are sought as chemical tools and potential therapeutics, such as for cancer. Activators are available for Sirt1 and exploit its unique N-terminus, whereas drug-like activators for Sirt2–7 are lacking. We synthesized and screened pyrrolo[1,2-a]quinoxaline derivatives, yielding the first synthetic Sirt6 activators. Biochemical assays show direct, substrate-independent compound binding to the Sirt6 catalytic core and potent activation of Sirt6-dependent deacetylation of peptide substrates and complete nucleosomes. Crystal structures of Sirt6/activator complexes reveal that the compounds bind to a Sirt6-specific acyl channel pocket and identify key interactions. Our results establish potent Sirt6 activation with small molecules and provide a structural basis for further development of Sirt6 activators as tools and therapeutics.
Co-reporter:Clemens Steegborn
Journal of Structural Biology 2017 Volume 198, Issue 1(Issue 1) pp:
Publication Date(Web):1 April 2017
DOI:10.1016/j.jsb.2017.02.010
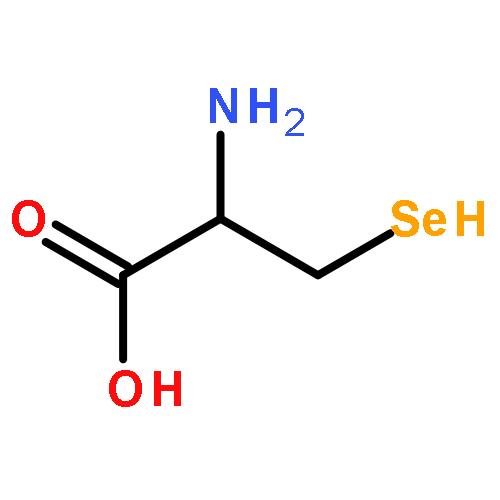
Co-reporter:Ulrich Schweizer, Holly Towell, Allegra Vit, Alfonso Rodriguez-Ruiz, Clemens Steegborn
Molecular and Cellular Endocrinology 2017 Volume 458(Volume 458) pp:
Publication Date(Web):15 December 2017
DOI:10.1016/j.mce.2017.01.026
•Iodothyronine hormones and derivatives bind to many evolutionary unrelated proteins.•We describe iodothyronine binding sites and ligand binding modes of these proteins.•Physiological and synthetic compounds can interfere with iodothyronine binding.Thyroid hormones and their metabolites constitute a vast class of related iodothyronine compounds that contribute to the regulation of metabolic activity and cell differentiation. They are in turn transported, transformed and recognized as signaling molecules through binding to a variety of proteins from a wide range of evolutionary unrelated protein families, which renders these proteins and their iodothyronine binding sites an example for extensive convergent evolution. In this review, we will briefly summarize what is known about iodothyronine binding sites in proteins, the modes of protein/iodothyronine interaction, and the ligand conformations. We will then discuss physiological and synthetic compounds, including popular drugs and food components, that can interfere with iodothyronine binding and recognition by these proteins. The discussion also includes compounds persisting in the environment and acting as endocrine disrupting chemicals.Download high-res image (205KB)Download full-size image
Co-reporter:Melanie Gertz
Cellular and Molecular Life Sciences 2016 Volume 73( Issue 15) pp:2871-2896
Publication Date(Web):2016 August
DOI:10.1007/s00018-016-2180-7
Sirtuins are an evolutionary conserved family of NAD+-dependent protein lysine deacylases. Mammals have seven Sirtuin isoforms, Sirt1–7. They contribute to regulation of metabolism, stress responses, and aging processes, and are considered therapeutic targets for metabolic and aging-related diseases. While initial studies were focused on Sirt1 and 2, recent progress on the mitochondrial Sirtuins Sirt3, 4, and 5 has stimulated research and drug development for these isoforms. Here we review the roles of Sirtuins in regulating mitochondrial functions, with a focus on the mitochondrially located isoforms, and on their contributions to disease pathologies. We further summarize the compounds available for modulating the activity of these Sirtuins, again with a focus on mitochondrial isoforms, and we describe recent results important for the further improvement of compounds. This overview illustrates the potential of mitochondrial Sirtuins as drug targets and summarizes the status, progress, and challenges in developing small molecule compounds modulating their activity.
Co-reporter:Joop van den Heuvel;Sebastien Moniot;Jochen Buck;Lonny R. Levin;Silke Kleinboelting;Michael Weyand;Ana Diaz
PNAS 2014 Volume 111 (Issue 10 ) pp:3727-3732
Publication Date(Web):2014-03-11
DOI:10.1073/pnas.1322778111
cAMP is an evolutionary conserved, prototypic second messenger regulating numerous cellular functions. In mammals, cAMP is
synthesized by one of 10 homologous adenylyl cyclases (ACs): nine transmembrane enzymes and one soluble AC (sAC). Among these,
only sAC is directly activated by bicarbonate (HCO3−); it thereby serves as a cellular sensor for HCO3−, carbon dioxide (CO2), and pH in physiological functions, such as sperm activation, aqueous humor formation, and metabolic regulation. Here, we
describe crystal structures of human sAC catalytic domains in the apo state and in complex with substrate analog, products,
and regulators. The activator HCO3− binds adjacent to Arg176, which acts as a switch that enables formation of the catalytic cation sites. An anionic inhibitor,
4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid, inhibits sAC through binding to the active site entrance, which blocks
HCO3− activation through steric hindrance and trapping of the Arg176 side chain. Finally, product complexes reveal small, local
rearrangements that facilitate catalysis. Our results provide a molecular mechanism for sAC catalysis and cellular HCO3− sensing and a basis for targeting this system with drugs.
Co-reporter:Ulrich Schweizer;Christine Schlicker;Doreen Braun;Josef Köhrle
PNAS 2014 Volume 111 (Issue 29 ) pp:10526-10531
Publication Date(Web):2014-07-22
DOI:10.1073/pnas.1323873111
Local levels of active thyroid hormone (3,3′,5-triiodothyronine) are controlled by the action of activating and inactivating
iodothyronine deiodinase enzymes. Deiodinases are selenocysteine-dependent membrane proteins catalyzing the reductive elimination
of iodide from iodothyronines through a poorly understood mechanism. We solved the crystal structure of the catalytic domain
of mouse deiodinase 3 (Dio3), which reveals a close structural similarity to atypical 2-Cys peroxiredoxin(s) (Prx). The structure
suggests a route for proton transfer to the substrate during deiodination and a Prx-related mechanism for subsequent recycling
of the transiently oxidized enzyme. The proposed mechanism is supported by biochemical experiments and is consistent with
the effects of mutations of conserved amino acids on Dio3 activity. Thioredoxin and glutaredoxin reduce the oxidized Dio3
at physiological concentrations, and dimerization appears to activate the enzyme by displacing an autoinhibitory loop from
the iodothyronine binding site. Deiodinases apparently evolved from the ubiquitous Prx scaffold, and their structure and catalytic
mechanism reconcile a plethora of partly conflicting data reported for these enzymes.
Co-reporter:Michael H. Suhre;Thomas Scheibel, ;Melanie Gertz
Acta Crystallographica Section F 2014 Volume 70( Issue 6) pp:769-772
Publication Date(Web):
DOI:10.1107/S2053230X14006165
In order to deal with the dynamic ocean environment, blue mussels adhere to various surfaces via their collagenous byssal threads. PTMP1 (proximal thread matrix protein 1) is one identified matrix protein residing in the proximal thread and is capable of collagen binding. Its sequence comprises two von Willebrand factor type A-like repeats. In order to characterize the structure and domain architecture of PTMP1, recombinant protein was crystallized by vapour diffusion. The obtained crystals diffracted to 1.95 Å resolution and belonged to space group P21, with unit-cell parameters a = 62.0, b = 62.3, c = 122.6 Å, β = 102.2°. The Matthews coefficient suggested the presence of two monomers in the asymmetric unit and 48.3% solvent content.
Co-reporter:Silke Kleinboelting;Joop van den Heuvel;Christian Kambach;Michael Wey;Martina Leipelt
Acta Crystallographica Section F 2014 Volume 70( Issue 4) pp:467-469
Publication Date(Web):
DOI:10.1107/S2053230X14004014
The second messenger cAMP is synthesized in mammals by ten differently regulated adenylyl cyclases (AC1–10). These ACs are grouped into nucleotidyl cyclase class III based on homologies in their catalytic domains. The catalytic domain of AC10 is unique, however, in being activated through direct interaction with calcium and bicarbonate. Here, the production, crystallization and X-ray diffraction analysis of the catalytic domain of human AC10 are described as a basis for structural studies of regulator binding sites and mechanisms. The recombinant protein had high specific AC activity, and crystals of AC10 in space group P63 diffracted to ∼2.0 Å resolution on a synchrotron beamline. A complete diffraction data set revealed unit-cell parameters a = b = 99.65, c = 98.04 Å, indicating one AC10 catalytic domain per asymmetric unit, and confirmed that the obtained crystals are suitable for structure solution and mechanistic studies.
Co-reporter:Giang Thi Tuyet Nguyen, Melanie Gertz, Clemens Steegborn
Chemistry & Biology 2013 Volume 20(Issue 11) pp:1375-1385
Publication Date(Web):21 November 2013
DOI:10.1016/j.chembiol.2013.09.019
•4′-bromo-resveratrol is a potent inhibitor of human Sirt1 and Sirt3•Crystal structures of Sirt3/peptide complexes reveal two compound binding sites•4′-bromo-resveratrol inhibits by binding to C-site and another active site pocket•A binding site on the Sirtuin surface is a candidate site for allosteric activatorsSirtuins are protein deacetylases regulating aging processes and various physiological functions. Resveratrol, a polyphenol found in red wine, activates human Sirt1 and inhibits Sirt3, and it can mimic calorie restriction effects, such as lifespan extension in lower organisms. The mechanism of Sirtuin modulation by resveratrol is not well understood. We used 4′-bromo-resveratrol (5-(2-(4-hydroxyphenyl)vinyl)-1,3-benzenediol) to study Sirt1 and Sirt3 modulation. Despite its similarity to the Sirt1 activator resveratrol, the compound potently inhibited both, Sirt1 and Sirt3. Crystal structures of Sirt3 in complex with a fluorophore-labeled and with a native substrate peptide, respectively, in presence of 4′-bromo-resveratrol reveal two compound binding sites. Biochemical studies identify the internal site and substrate competition as the mechanism for inhibition, providing a drug target site, and homology modeling suggests that the second, allosteric site might indicate the site for Sirt1 activation.Figure optionsDownload full-size imageDownload high-quality image (246 K)Download as PowerPoint slide
Co-reporter:Benjamin Suenkel, Frank Fischer, Clemens Steegborn
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 1) pp:143-146
Publication Date(Web):1 January 2013
DOI:10.1016/j.bmcl.2012.10.136
Sirtuins are NAD+ consuming protein deacylases involved in many cellular processes from DNA-repair to metabolism. Their contribution to age-related and metabolic diseases makes them attractive pharmaceutical targets. Few pharmacological inhibitors have been reported yet for human Sirt5 since substrates and assays for reliable testing of its activity were unavailable until recently, and most modulators of other Sirtuins were not tested against Sirt5 and therefore have only partially characterized isoform selectivities. We used here improved substrates and assays for testing of known Sirtuin inhibitors for their effects on two activities of human Sirt5, the generic Sirtuin activity deacetylation and the more pronounced Sirt5 activity desuccinylation. Our tests show that most of the compounds have no significant effect on either Sirt5 activity. The indole GW5074, however, was found to be a potent inhibitor for Sirt5’s desuccinylation activity, identifying a first pharmacological scaffold for development into Sirt5-specific inhibitors. Interestingly, the compound showed weaker effects in Sirt5 deacetylation assays and also varying potencies against different peptide sequences, indicating a substrate-specific effect of GW5074.
Co-reporter:Melanie Gertz;Frank Fischer;Giang Thi Tuyet Nguyen;Mike Schutkowski;Michael Weyand;Mahadevan Lakshminarasimhan
PNAS 2013 Volume 110 (Issue 30 ) pp:E2772-E2781
Publication Date(Web):2013-07-23
DOI:10.1073/pnas.1303628110
Sirtuins are protein deacetylases regulating metabolism and stress responses. The seven human Sirtuins (Sirt1–7) are attractive
drug targets, but Sirtuin inhibition mechanisms are mostly unidentified. We report the molecular mechanism of Sirtuin inhibition
by 6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide (Ex-527). Inhibitor binding to potently inhibited Sirt1 and Thermotoga maritima Sir2 and to moderately inhibited Sirt3 requires NAD+, alone or together with acetylpeptide. Crystal structures of several Sirtuin inhibitor complexes show that Ex-527 occupies
the nicotinamide site and a neighboring pocket and contacts the ribose of NAD+ or of the coproduct 2’-O-acetyl-ADP ribose. Complex structures with native alkylimidate and thio-analog support its catalytic relevance and show,
together with biochemical assays, that only the coproduct complex is relevant for inhibition by Ex-527, which stabilizes the
closed enzyme conformation preventing product release. Ex-527 inhibition thus exploits Sirtuin catalysis, and kinetic isoform
differences explain its selectivity. Our results provide insights in Sirtuin catalysis and inhibition with important implications
for drug development.
Co-reporter:Sébastien Moniot, Mike Schutkowski, Clemens Steegborn
Journal of Structural Biology (May 2013) Volume 182(Issue 2) pp:136-143
Publication Date(Web):1 May 2013
DOI:10.1016/j.jsb.2013.02.012
Sirtuins are NAD+-dependent protein deacetylases that regulate metabolism and aging-related processes. Sirt2 is the only cytoplasmic isoform among the seven mamalian Sirtuins (Sirt1-7) and structural information concerning this isoform is limited. We crystallized Sirt2 in complex with a product analog, ADP-ribose, and solved this first crystal structure of a Sirt2 ligand complex at 2.3 Å resolution. Additionally, we re-refined the structure of the Sirt2 apoform and analyzed the conformational changes associated with ligand binding to derive insights into the dynamics of the enzyme. Our analyses also provide information on Sirt2 peptide substrate binding and structural states of a Sirt2-specific protein region, and our insights and the novel Sirt2 crystal form provide helpful tools for the development of Sirt2 specific inhibitors.
Co-reporter:Hüsnü Topal, Nanette B. Fulcher, Jacob Bitterman, Eric Salazar, ... Clemens Steegborn
Journal of Molecular Biology (17 February 2012) Volume 416(Issue 2) pp:271-286
Publication Date(Web):17 February 2012
DOI:10.1016/j.jmb.2011.12.045
Pseudomonas aeruginosa is an opportunistic bacterial pathogen and a major cause of healthcare-associated infections. While the organism's intrinsic and acquired resistance to most antibiotics hinders treatment of P. aeruginosa infections, the regulatory networks controlling its virulence provide novel targets for drug development. CyaB, a key regulator of P. aeruginosa virulence, belongs to the Class III adenylyl cyclase (AC) family of enzymes that synthesize the second messenger cyclic adenosine 3′,5′-monophosphate. These enzymes consist of a conserved catalytic domain fused to one or more regulatory domains. We describe here the biochemical and structural characterization of CyaB and its inhibition by small molecules. We show that CyaB belongs to the Class IIIb subfamily, and like other subfamily members, its activity is stimulated by inorganic carbon. CyaB is also regulated by its N-terminal MASE2 (membrane-associated sensor 2) domain, which acts as a membrane anchor. Using a genetic screen, we identified activating mutations in CyaB. By solving the crystal structure of the CyaB catalytic domain, we rationalized the effects of these mutations and propose that CyaB employs regulatory mechanisms similar to other Class III ACs. The CyaB structure further indicates subtle differences compared to other Class III ACs in both the active site and the inhibitor binding pocket. Consistent with these differences, we observed a unique inhibition profile, including identification of a CyaB selective compound. Overall, our results reveal mechanistic details of the physiological and pharmacological regulation of CyaB and provide the basis for its exploitation as a therapeutic drug target.Download high-res image (154KB)Download full-size imageHighlights► Characterization of CyaB, an AC regulating P. aeruginosa virulence. ► The MASE2 domain of CyaB acts as membrane anchor and influences cyclase activity. ► Residues involved in CyaB regulation identified using a random mutagenesis screen. ► A CyaB catalytic core crystal structure reveals location and function of the residues. ► Modulation of CyaB through CO2/HCO3− and known AC inhibitors