Co-reporter:Zu-xu Chen, Hua Zhong, Hai-tao Yu
Computational and Theoretical Chemistry 2017 Volume 1099(Volume 1099) pp:
Publication Date(Web):1 January 2017
DOI:10.1016/j.comptc.2016.11.006
•The NH3-loss pathway of [Asp-H]− is dominant as the temperature is lower than 719 K.•The CO2-loss is the kinetically most favorable as the temperature is higher than 719 K.•The H2O-loss pathway is less favorable than the NH3- and CO2-loss channels.In this study, the fragmentation processes of deprotonated aspartic acid to eliminate CO2, NH3, and H2O were investigated by a quantum-mechanism computation at the B3LYP/6-311++G(2df,2pd) and MP2/6-31+G(d,p) levels of theory. By constructing the fragmentation reaction potential energy profile using the Gibbs free energies and enthalpies of the located stationary points, we explored both the preferred dissociation pathways and the product distribution. Furthermore, the thermal energy correction to key stationary points was computed to evaluate the temperature dependences of the dominant reaction channels and product distribution. Additionally, the similarities and differences between the present theoretical investigation and available experiment were discussed in detail.Fragmentation pathways and product distribution of deprotonated aspartic acid were investigated.Download high-res image (159KB)Download full-size image
Co-reporter:Hong Chen, Hai-Tao Yu, Ying Xie
Materials Chemistry and Physics 2016 Volume 174() pp:195-203
Publication Date(Web):1 May 2016
DOI:10.1016/j.matchemphys.2016.02.075
•The O- and O2-terminated (110) surfaces of SrHfO3 can be stabilized in some special regions.•No (001) terminations of SrHfO3 can be stabilized.•The AFD relaxation results in the large rotation displacements of terminations of SrHfO3.A first-principles thermodynamics characterization of the stabilities and structures of SrHfO3 (110) polar terminations has been performed using density functional theory simulations and the surface grand potential technique. The investigated terminations include the SrHfO-, O2-, HfO-, Sr-, and O-terminated (110) surfaces. Based on the computed surface relaxations (reconstructions), charge redistribution, and electronic structures, the polarity compensation mechanisms of the terminations, i.e., how the polarity compensation criterion is fulfilled, are discussed in detail. To account for the effect of the chemical environment and make a reasonable comparison, the (001) surface and the precipitation of small mono-metal oxide crystals on the grown surfaces are considered in the construction of the stability diagram. Based on the constructed stability phase diagram, the stability regions of the (110) and (001) terminations are determined. Additionally, a comparison of the stability behaviors with several analogues is further drawn.
Co-reporter:Bing Zheng, Hai-tao Yu, Yong-fu Lian, Ying Xie
Chemical Physics Letters 2016 Volume 648() pp:81-86
Publication Date(Web):16 March 2016
DOI:10.1016/j.cplett.2016.01.074
•New boron α6- and β14-sheets were constructed from B36 and B35 motifs, respectively.•Binding energies and MD simulations show they are thermodynamically stable.•Their work functions are the highest among MTH sheets and very close to graphene.In this study, we report the quantum-mechanical characterization of two novel α- and β-type 2D pure boron sheets, i.e., α6- and β14-sheets, constructed from the experimentally available B36 and B35 building blocks. Ten isomeric configurations were located. Using the calculated binding energies, the thermodynamic stability of these structures was considered in detail. Additionally, we calculated the work functions of α6- and β14-sheets. The results clearly demonstrate that their work functions (approximately 4.6 eV) are the highest among all of the reported mixed triangular-hexagonal type 2D boron sheets and are very similar to that of graphene.
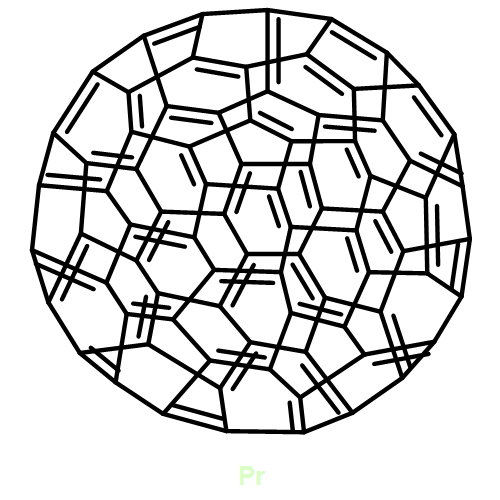
Co-reporter:Yan-li Zhao, Qin Zhou, Yong-fu Lian, Hai-tao Yu
Diamond and Related Materials 2016 Volume 64() pp:110-118
Publication Date(Web):April 2016
DOI:10.1016/j.diamond.2016.02.004
•Pr@D3h(14246)-C74(C6H3Cl2) was prepared and isolated.•MS and UV–vis-NIR spectroscopy of Pr@D3h(14246)-C74(C6H3Cl2) are available.•Structures of Pr@D3h(14246)-C74(C6H3Cl2) and Pr@D3h(14246)-C74 were determined.Pr@C74(C6H3Cl2), a novel dichlorophenyl-functionalized derivative of the Pr-containing monometallofullerene Pr@C74, was successfully prepared and isolated. Its molecular structure was determined as Pr@D3h(14246)-C74(C6H3Cl2) by a combined mass spectrometry (MS), UV–visible-near-infrared (UV–vis-NIR) absorption spectroscopy, and quantum-mechanics characterization. Using the computed theoretical data, we evaluated the isomeric structures, metal-to-cage electron transfer, and metal-cage interactions of Pr@C74(C6H3Cl2) and the pristine Pr@C74. Furthermore, the relative reactivity of C74, Pr@C74, and Pr@C74(C6H3Cl2) was estimated by the calculated electronic structures and band gaps, and the results indicated that the open-shell structure and relatively small gap should be responsible for the unsuccessful isolation of C74 and Pr@C74.Prime novelty statementWe successfully prepared and isolated a novel Pr-based monometallofullerene derivative, Pr@C74(C6H3Cl2), for the first time. Its structure, together with the nonderivatized Pr@C74, was discussed in detail based on a combined UV–visible-near-infrared and quantum-mechanics characterization.
Co-reporter:Fazal Raziq
The Journal of Physical Chemistry C 2016 Volume 120(Issue 1) pp:98-107
Publication Date(Web):December 8, 2015
DOI:10.1021/acs.jpcc.5b10313
We have successfully synthesized boron-doped g-C3N4 nanosheets (B-CN) and its nanocomposites with nanocrystalline anatase TiO2 (T/B-CN). The as-prepared T/B-CN nanocomposites with the proper amounts of boron and TiO2 exhibit rather high cocatalyst-free photoactivities for producing H2 from CH3OH solution (∼29× higher) and CH4 from CO2-containing water (∼16× higher) under visible-light irradiation, compared to those of bare g-C3N4. This is attributed to the greatly enhanced photogenerated charge separation after doping boron and subsequent coupling with TiO2, mainly based on the measurements of atmosphere-controlled steady-state surface photovoltage spectra, transient-state surface photovoltage responses, photoluminescence spectra, and fluorescence spectra related to the produced hydroxyl radical amount. It is suggested for the first time that the great charge separation enhancement results from the B-induced surface states near the valence band top to trap holes and the formed heterojunctions to transfer electrons from B-CN to TiO2. Moreover, the created surface states are also responsible for the visible-light extension from 450 nm of g-C3N4 to 500 nm of B-CN (T/B-CN) for solar fuel production. Interestingly, the obtained 6T/6B-CN exhibits much larger quantum efficiencies, which are 3.08% for hydrogen evolution and 1.68% for CH4 production at λ = 420 nm, respectively, with 5.1× and 7.6× enhancement as compared to CN, even superior to other works. This work will provide feasible routes to synthesize g-C3N4-based nanophotocatalysts for efficient solar fuel production.
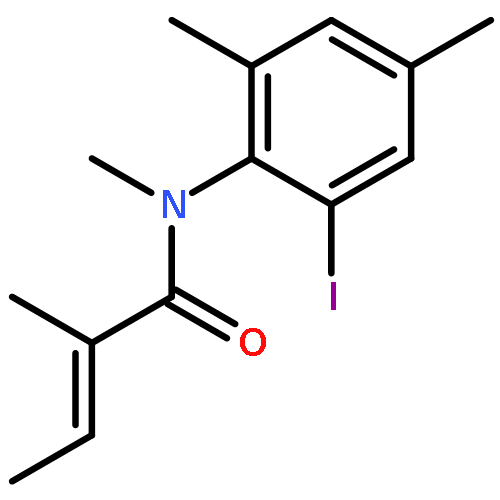
Co-reporter:Bai-jian Li, Hua Zhong, and Hai-tao Yu
The Journal of Physical Chemistry B 2016 Volume 120(Issue 50) pp:12950-12958
Publication Date(Web):November 28, 2016
DOI:10.1021/acs.jpcb.6b10344
In this study, we employed the density functional method to simulate AIBN/HSnBu3-mediated radical cyclizations with different axially chiral conformers of N-(2-iodo-4,6-dimethylphenyl)-N,2-dimethyl-(2E)-butenamide as substrates. We constructed a reaction potential energy profile using the Gibbs free energies of the located stationary points. The thermodynamic and kinetic data of the profile were further used to evaluate the regioselectivity, stereoselectivity, and product distribution of the cyclizations. Additionally, we compared the present HSnBu3-mediated radical cyclization with the experimentally available Heck reaction and found that such a radical cyclization can convert (M,Z) and (P,Z) o-iodoanilide substrates to centrally chiral products with high chirality transfer. The goal of this study was to estimate the practicality of theoretically predicting the memory of chirality in such radical cyclizations. The present results can provide a strategy from a theoretical viewpoint for experimentally synthesizing highly stereoselective carbocyclic and heterocyclic compounds using radical cyclization methods.
Co-reporter:Yan-li Zhao, Qin Zhou, Yong-fu Lian and Hai-tao Yu
RSC Advances 2015 vol. 5(Issue 118) pp:97568-97578
Publication Date(Web):09 Nov 2015
DOI:10.1039/C5RA17608E
A novel Pr-based monometallofullerene derivative, Pr@C72(C6H3Cl2), was successfully prepared and isolated. Its molecular composition was determined by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. The molecular structure of Pr@C72(C6H3Cl2) was verified as Pr@C2(10612)-C72(C6H3Cl2) by combined UV-visible-near-infrared absorption spectroscopy and quantum mechanics characterization, as well as a comparison with the structurally characterized analogue La@C72(C6H3Cl2). Furthermore, an additional computation indicated that the nonderivatized Pr@C72 has the lowest-lying structure of Pr@C2(10612)-C72, followed by Pr@C2v(11188)-C72, which lies only 0.62 kcal mol−1 above Pr@C2(10612)-C72. In addition, the temperature dependence of their thermodynamic distribution was estimated. We also analyzed the charge transfer and orbital interaction between the endohedral Pr atom and the carbon cage as well as the electronic configuration and formal charge state of the encapsulated Pr atom based on the computed quantum mechanics data.
Co-reporter:Bing Zheng, Hai-tao Yu, Ying Xie, and Yong-fu Lian
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 22) pp:19690
Publication Date(Web):October 21, 2014
DOI:10.1021/am504674p
First-principles density functional theory calculations were performed to study the effect of Li adsorption on the structural and electronic properties, particularly the work function, of boron α-sheet. The calculated binding energies indicated that boron α-sheet could be well stabilized by the adsorption of Li atoms. Furthermore, the work functions of Li-adsorbed boron α-sheets were observed to decrease drastically with increasing Li coverage. The work functions are lower than that of Mg and even, for some of them, lower than that of Ca, indicating a considerable potential application of Li-adsorbed boron α-sheets as field-emission and electrode materials. Based on the calculated geometric and electronic structures, we discuss in details some possible aspects affecting the work function. The Li coverage dependence of the work functions of Li-adsorbed boron α-sheets was further confirmed by electrostatic potential analyses. The relationship between the work function variation and the Fermi and vacuum energy level shifts was also discussed, and we observed that the variation of the work function is primarily associated with the shift of the Fermi energy level. It is the surface dipole formed by the interaction between adatoms and substrate that should be responsible for the observed variation of the work function, whereas the increasing negative charge and rumpling for boron α-sheet only play minor roles. Additionally, the effect of Li adatoms on the work function of boron α-sheet was confirmed to be much stronger than that of graphene or a graphene double layer.Keywords: binding energy; buckled boron α-sheet; electronic population; electronic structure; electrostatic potential; work function
Co-reporter:Ying Xie;Haitao Yu
Chemical Research in Chinese Universities 2014 Volume 30( Issue 5) pp:794-799
Publication Date(Web):2014 October
DOI:10.1007/s40242-014-4174-z
The band structures, electron density differences, and surface energies of five different BaTiO3 (110) terminations were investigated by first-principles calculations. According to the calculated results of electron density differences, the bonding characteristics of these considered terminations were discussed. The computational results indicate that the BaTiO-terminated surface is metallic, while the O2-, O-, Ba- and TiO-terminated surfaces are all insulative. Furthermore, the computed surface energies suggest that for the considered terminations, the polarity compensation achieved through surface reconstruction or surface defect is more effective than by change in surface electronic structure. The defected or reconstructed terminations predominate over cleavage and construction of BaTiO3 crystal along (110) direction.
Co-reporter:Ying Wang, Hua Zhong, Hai-tao Yu
Computational and Theoretical Chemistry (1 March 2017) Volume 1103() pp:
Publication Date(Web):1 March 2017
DOI:10.1016/j.comptc.2017.01.018
•The molecular fragment [L–(2H,O)]+[L–(2H,O)]+ (L = 5-methoxyflavone) can be formed by the loss of H2O.•The fragmentation to [L–(H,C,O)]+[L–(H,C,O)]+ is via the loss of HCO to give conformers 18 and 19.•The favorable fragmentation product [L–(2H,C,2O)]+[L–(2H,C,2O)]+ is the ion-structure 35, not experimentally suggested 4.In this study, density functional theory calculations were employed to investigate the dissociation of the molecular ion [L]+[L]+ (L = 5-methoxyflavone). Using the Gibbs free energies of the optimized stationary points, [L]+[L]+ dissociation potential energy profiles were constructed, and the dissociation mechanism, including the detailed fragmentation pathways to [L–(2H,O)]+[L–(2H,O)]+, [L−(H,C,O)]+ and [L–(2H,C,2O)]+[L–(2H,C,2O)]+; fragment configurations; and product distribution, was further explored. Finally, the similarities and differences between the theoretical and available experimental results were discussed in detail. This study provides new insight into the dissociation of flavonoids and their analogues in mass spectrometry.In this study, we used quantum chemical calculations to determine the detailed dissociation pathways of the 5-methoxyflavone radical cation and the corresponding product structures.