Co-reporter:Geetu Sharma, Sergey V. Ushakov, Hui Li, Ricardo H. R. Castro, and Alexandra Navrotsky
The Journal of Physical Chemistry C May 18, 2017 Volume 121(Issue 19) pp:10392-10392
Publication Date(Web):May 2, 2017
DOI:10.1021/acs.jpcc.7b01262
Thermodynamics of nanomaterials is strongly influenced by the energetic contribution from atoms located at the interfaces. Therefore, accurately assessing the surface energy of nanomaterials is essential for calculating and predicting thermodynamic properties. In the present work, surface energy of amorphous hafnium oxide (am-HfO2) nanoparticles was measured using independent calorimetric techniques. am-HfO2 nanoparticles were synthesized by condensation from a gas phase generated through laser evaporation of bulk HfO2 targets at 0.1 Torr oxygen pressure. Their surface energy was directly measured using high-temperature oxide melt solution calorimetry, differential scanning calorimetry, and water adsorption calorimetry. The measured surface energies using the above techniques, respectively, are 0.76 ± 0.12, 0.47 ± 0.2, and 0.59 ± 0.1 J/m2. The differences among the surface energy values are about 0.3 J/m2, which is generally within the experimental uncertainties and different assumptions for each technique. The surface energy of the amorphous phase is substantially smaller than that of crystalline phases, as seen previously for other oxides. Thus, the amorphous phase may be thermodynamically favored when small particles are produced and retained.
Co-reporter:Zamirbek Akimbekov, Athanassios D. Katsenis, G. P. Nagabhushana, Ghada Ayoub, Mihails Arhangelskis, Andrew J. Morris, Tomislav Friščić, and Alexandra Navrotsky
Journal of the American Chemical Society June 14, 2017 Volume 139(Issue 23) pp:7952-7952
Publication Date(Web):May 18, 2017
DOI:10.1021/jacs.7b03144
We provide the first combined experimental and theoretical evaluation of how differences in ligand structure and framework topology affect the relative stabilities of isocompositional (i.e., true polymorph) metal–organic frameworks (MOFs). We used solution calorimetry and periodic DFT calculations to analyze the thermodynamics of two families of topologically distinct polymorphs of zinc zeolitic imidazolate frameworks (ZIFs) based on 2-methyl- and 2-ethylimidazolate linkers, demonstrating a correlation between measured thermodynamic stability and density, and a pronounced effect of the ligand substituent on their stability. The results show that mechanochemical syntheses and transformations of ZIFs are consistent with Ostwald’s rule of stages and proceed toward thermodynamically increasingly stable, more dense phases.
Co-reporter:Geetu Sharma, Elayaraja Muthuswamy, Michael Naguib, Yury Gogotsi, Alexandra Navrotsky, and Di Wu
The Journal of Physical Chemistry C July 20, 2017 Volume 121(Issue 28) pp:15145-15145
Publication Date(Web):June 21, 2017
DOI:10.1021/acs.jpcc.7b02419
Intercalation of ions in layered materials has been explored to improve the rate capability in Li-ion batteries and supercapacitors. This work investigates the energetics of alkali ion exchange in a clay-like MXene, Ti3C2Tx, where Tx stands for anionic surface moieties, by immersion calorimetry in aqueous solutions. The measured immersion enthalpies of clay-like Ti3C2Tx, ΔHimm, at 25 °C in 1 M KCl, 1 M NaCl, 1 M LiCl, and nanopure water are −9.19 (±0.56), −5.90 (±0.31), −1.31 (±0.20), and −1.29 (±0.13) kJ/mol of MXene, respectively. Inductively coupled plasma mass spectrometry is used to obtain the concentrations of alkali ions in the solid and aqueous phases. Using these concentrations, the enthalpies of exchange of alkali metal ions (Li+, Na+, and K+) are calculated; ΔHex in 1 M KCl, 1 M NaCl, 1 M LiCl, and nanopure water are −9.3 (±2.2), 21.0 (±0.9), −1.3 (±0.2), and 302.4 (±0.6) kJ/mol of MXene, respectively. Both immersion and exchange enthalpies are most exothermic for potassium. This suggests that K+ ions interact more strongly with anions present in the interlayers of this MXene than Na+ and Li+ ions. Water vapor adsorption calorimetry indicates very weak interaction of water with the MXene, while immersion calorimetry suggests a weakly hydrophilic nature of the MXene surface.
Journal of Materials Chemistry A 2017 vol. 5(Issue 25) pp:12951-12957
Publication Date(Web):2017/06/27
DOI:10.1039/C7TA02434G
Perovskite-structured lithium lanthanum titanate (LLTO) Li3xLa0.67−xTiO3 (compositions x = 0.04 to 0.15) has been prepared by conventional solid state reaction. The phase purity and crystal structural changes were investigated with XRD, FTIR and Raman. The vibrational spectra reveal the interaction between metal cation and oxygen anion with increasing Li doping and structural evolution. LLTO and component oxides were studied by high temperature oxide melt solution calorimetry. The formation enthalpies of LLTO from oxides are exothermic for all compositions, indicating thermodynamic stability. There are two regimes in the trend of formation enthalpy with increasing Li concentration. In the first regime, x ≤0.08, the formation enthalpies vary slowly with composition, but the lowest stability by about 1.5 kJ mol−1 is seen at x = 0.06. An abrupt change in the formation enthalpy trend is observed in the second regime when x ≥0.1, where maximum lithium ion conductivity (at x = 0.10) is reported. The least stable composition, x = 0.06, occurs where maximum charge carrier concentration and lowest activation energy is reported. From the thermodynamic study, it is clear that the energetically least stable composition correlates with lowest activation energy whereas the sharp change in formation enthalpy trend correlates with highest Li-ion conductivity.
Co-reporter:Lei Zhang, Anna Shelyug, Alexandra Navrotsky
The Journal of Chemical Thermodynamics 2017 Volume 114(Volume 114) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.jct.2017.05.026
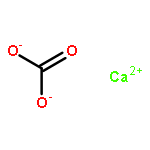
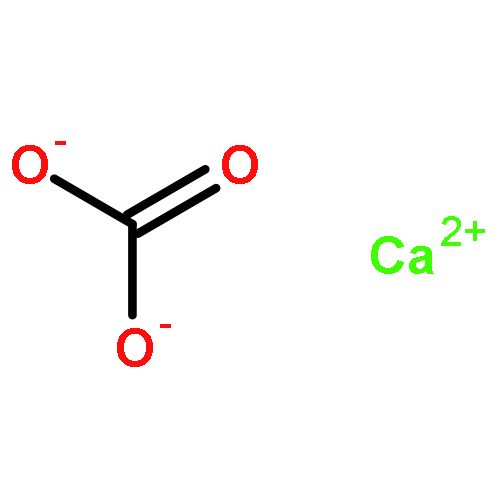
•Enthalpies of mixing of UO2 – ThO2 and UO2 – ZrO2 systems have been measured.•Volume mismatch linearly depends on interaction parameter in related systems.•UO2 – ZrO2 is an exception to this correlation showing zero heat of mixing.The enthalpies of formation of cubic urania – thoria (c-ThxU1−xO2+y) and urania – zirconia (c-ZrxU1−xO2, x < 0.3) solid solutions at 25 °C from end-member binary oxides (c-UO2, and c-ThO2 or m-ZrO2) have been measured by high temperature oxide melt solution calorimetry. The enthalpies of mixing for both systems are zero within experimental error. The interaction parameters for binary solid solutions MO2 – M′O2 (M, M′ = U, Th, Ce, Zr, and Hf), fitted by regular and subregular thermodynamic models using both calorimetric and computational data, increase linearly with the corresponding volume mismatch. Cubic UO2 – ZrO2 appears to be an exception to this correlation and shows a zero heat of mixing despite large size mismatch, suggestive of some short-range ordering and/or incipient phase separation to mitigate the strain. The incorporation of ZrO2 into UO2 stabilizes the system and makes it a potential candidate for immobilization and disposal of nuclear waste.
Co-reporter:Jacob Schliesser, Kristina Lilova, Eric M. Pierce, Lili Wu, David M. Missimer, Brian F. Woodfield, Alexandra Navrotsky
The Journal of Chemical Thermodynamics 2017 Volume 114(Volume 114) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.jct.2017.05.035
•Heat capacities of sulfate, perrhenate, chloride, and iodide sodalites were measured from 2 K to 300 K.•Heat capacities were fitted to equations in three temperature ranges.•Standard entropies and Gibbs energies were calculated.•All four sodalites undergo a phase transition below room temperature for which thermodynamic parameters were determined.•This work is relevant to nuclear waste disposal.Heat capacities of sulfate, perrhenate, chloride, and iodide sodalites with the ideal formula Na8Al6Si6O24X2 (X = SO4, ReO4, Cl, I) were measured from 2 K to 300 K using a Quantum Design Physical Property Measurement System (PPMS). From the heat capacity data, the standard thermodynamic functions were determined. All four sodalites undergo a phase transition below room temperature for which thermodynamic parameters were determined. Additionally, the heat capacity of one of the constituent compounds (NaReO4) was measured.
Co-reporter:Sriram Goverapet Srinivasan;Radha Shivaramaiah;Paul R. C. Kent;Andrew G. Stack;Richard Riman;Andre Anderko;Vyacheslav S. Bryantsev
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 11) pp:7820-7832
Publication Date(Web):2017/03/15
DOI:10.1039/C7CP00811B
Bastnäsite, a fluoro-carbonate mineral, is the single largest mineral source of light rare earth elements (REE), La, Ce and Nd. Enhancing the efficiency of separation of the mineral from gangue through froth flotation is the first step towards meeting an ever increasing demand for REE. To design and evaluate collector molecules that selectively bind to bastnäsite, a fundamental understanding of the structure and surface properties of bastnäsite is essential. In our earlier work (J. Phys. Chem. C, 2016, 120, 16767), we carried out an extensive study of the structure, surface stability and water adsorption energies of La-bastnäsite. In this work, we make a comparative study of the surface properties of Ce-bastnäsite, La-bastnäsite, and calcite using a combination of density functional theory (DFT) and water adsorption calorimetry. Spin polarized DFT+U calculations show that the exchange interaction between the electrons in Ce 4f orbitals is negligible and that these orbitals do not participate in bonding with the oxygen atom of the adsorbed water molecule. In agreement with calorimetry, DFT calculations predict larger surface energies and stronger water adsorption energies on Ce-bastnäsite than on La-bastnäsite. The order of stabilities for stoichiometric surfaces is as follows: [100] > [101] > [102] > [0001] > [112] > [104] and the most favorable adsorption sites for water molecules are the same as for La-bastnäsite. In agreement with water adsorption calorimetry, at low coverage water molecules are strongly stabilized via coordination to the surface Ce3+ ions, whereas at higher coverage they are adsorbed less strongly via hydrogen bonding interaction with the surface anions. Due to similar water adsorption energies on bastnäsite [101] and calcite [104] surfaces, the design of collector molecules that selectively bind to bastnäsite over calcite must exploit the structural differences in the predominantly exposed facets of these minerals.
Co-reporter:Liangjie FuHuaming Yang, Yuehua Hu, Di Wu, Alexandra Navrotsky
Chemistry of Materials 2017 Volume 29(Issue 3) pp:
Publication Date(Web):January 8, 2017
DOI:10.1021/acs.chemmater.6b05041
Well crystallized gamma alumina (γ-Al2O3) with high thermal stability is as an important catalyst support. A series of first row transition metal (TM) doped aluminas with ordered mesoporous structures and homogeneous distribution of TM in the bulk structure has been synthesized by a one-pot method. The structures are studied by powder X-ray diffraction (XRD) and transmission electron microscopy (TEM), while the electronic properties are explored by X-ray photoelectron spectroscopy (XPS), valence band XPS, and UV–vis spectra. To explore the influence of TM dopants on atomistic properties (bond length, charge state, band edge, and redox properties) of γ-Al2O3, the cation distribution of TM dopants is studied in detail by combining experiments and density functional theory (DFT) calculations. The cooperative effect of TM dopants and intrinsic defects in γ-Al2O3 induces a doping mechanism distinct from that in other spinel oxides; the concentration of Al vacancies (VAl) decreases with increasing atomic number of the TM dopant as a result of charge compensation effects. Such variation could be used to tailor the properties and alter the reactivity of γ-Al2O3.
Co-reporter:Alfred Pavlik III, Sergey V. Ushakov, Alexandra Navrotsky, Chris J. Benmore, Richard J.K. Weber
Journal of Nuclear Materials 2017 Volume 495(Volume 495) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.jnucmat.2017.08.031
•Lu2O3 and Yb2O3 retain bixbyite-type structure in oxygen and argon to melting.•Thermal expansion is close to linear from room temperature to melting.•There is no indication of Bredig transition or transformation to hexagonal phase.Knowledge of thermal expansion and high temperature phase transformations is essential for prediction and interpretation of materials behavior under the extreme conditions of high temperature and intense radiation encountered in nuclear reactors. Structure and thermal expansion of Lu2O3 and Yb2O3 were studied in oxygen and argon atmospheres up to their melting temperatures using synchrotron X-ray diffraction on laser heated levitated samples. Both oxides retained the cubic bixbyite C-type structure in oxygen and argon to melting. In contrast to fluorite-type structures, the increase in the unit cell parameter of Yb2O3 and Lu2O3 with temperature is linear within experimental error from room temperature to the melting point, with mean thermal expansion coefficients (8.5 ± 0.6) · 10−6 K−1 and (7.7 ± 0.6) · 10−6 K−1, respectively. There is no indication of a superionic (Bredig) transition in the C-type structure or of a previously suggested Yb2O3 phase transformation to hexagonal phase prior to melting.Download high-res image (157KB)Download full-size image
Manganese oxides with layer and tunnel structures occur widely in nature and inspire technological applications. Having variable
compositions, these structures often are found as small particles (nanophases). This study explores, using experimental thermochemistry,
the role of composition, oxidation state, structure, and surface energy in the their thermodynamic stability. The measured
surface energies of cryptomelane, sodium birnessite, potassium birnessite and calcium birnessite are all significantly lower
than those of binary manganese oxides (Mn3O4, Mn2O3, and MnO2), consistent with added stabilization of the layer and tunnel structures at the nanoscale. Surface energies generally decrease
with decreasing average manganese oxidation state. A stabilizing enthalpy contribution arises from increasing counter-cation
content. The formation of cryptomelane from birnessite in contact with aqueous solution is favored by the removal of ions
from the layered phase. At large surface area, surface-energy differences make cryptomelane formation thermodynamically less
favorable than birnessite formation. In contrast, at small to moderate surface areas, bulk thermodynamics and the energetics
of the aqueous phase drive cryptomelane formation from birnessite, perhaps aided by oxidation-state differences. Transformation
among birnessite phases of increasing surface area favors compositions with lower surface energy. These quantitative thermodynamic
findings explain and support qualitative observations of phase-transformation patterns gathered from natural and synthetic
manganese oxides.
Co-reporter:Xiaofeng Guo, Stéphanie Szenknect, Adel Mesbah, Nicolas Clavier, Christophe Poinssot, Di Wu, Hongwu Xu, Nicolas Dacheux, Rodney C. Ewing, and Alexandra Navrotsky
Chemistry of Materials 2016 Volume 28(Issue 19) pp:7117
Publication Date(Web):September 14, 2016
DOI:10.1021/acs.chemmater.6b03346
High-temperature oxide melt solution calorimetric measurements were completed to determine the enthalpies of formation of the uranothorite, (USiO4)x–(ThSiO4)1–x, solid solution. Phase-pure samples with x values of 0, 0.11, 0.21, 0.35, 0.71, and 0.84 were prepared, purified, and characterized by powder X-ray diffraction, electron probe microanalysis, thermogravimetric analysis and differential scanning calorimetry coupled with in situ mass spectrometry, and high-temperature oxide melt solution calorimetry. This work confirms the energetic metastability of coffinite, USiO4, and U-rich intermediate silicate phases with respect to a mixture of binary oxides. However, variations in unit cell parameters and negative excess volumes of mixing, coupled with strongly exothermic enthalpies of mixing in the solid solution, suggest short-range cation ordering that can stabilize intermediate compositions, especially near x = 0.5.
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 15) pp:10116-10122
Publication Date(Web):10 Mar 2016
DOI:10.1039/C5CP07918G
Transition metal cations (Mn2+, Co2+, Cu2+, and Zn2+) containing zeolites A and Y were synthesized by ion exchange and their thermochemistry was investigated by differential scanning calorimetry and high temperature oxide melt solution calorimetry. The enthalpies of formation from oxides for Mn, Co, Cu, and Zn zeolites A range from 14.0 ± 1.3 to 67.6 ± 5.5 kJ mol−1 and those for Mn, Co, Cu, and Zn zeolites Y range from 8.0 ± 2.0 to 32.6 ± 1.8 kJ mol−1. All these zeolites are thus metastable with respect to oxide components and to other dense phases. The formation enthalpies of Mn and Zn exchanged zeolites A and Y are less endothermic than those of corresponding Co and Cu exchanged A and Y. These energetics are consistent with metal oxygen bond lengths and related to crystal field effects of transition metal ions. Similar thermodynamic trends have been seen in transition metal containing spinel, olivine and pyroxene materials. The enthalpies of exchange of transition metals in zeolites A and Y with sodium in aqueous solution are calculated and suggest that these zeolites could be reasonably effective sorbents for heavy metal waste.
Co-reporter:Zamirbek Akimbekov, Di Wu, Carl K. Brozek, Mircea Dincă and Alexandra Navrotsky
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 2) pp:1158-1162
Publication Date(Web):26 Nov 2015
DOI:10.1039/C5CP05370F
The inclusion of solvent in metal–organic framework (MOF) materials is a highly specific form of guest–host interaction. In this work, the energetics of solvent MOF-5 interactions has been investigated by solution calorimetry in 5 M sodium hydroxide (NaOH) at room temperature. Solution calorimetric measurement of enthalpy of formation (ΔHf) of Zn4O(C8H4O4)3·C3H7NO (MOF-5·DMF) and Zn4O(C8H4O4)3·0.60C5H11NO (MOF-5·0.60DEF) from the dense components zinc oxide (ZnO), 1,4-benzenedicarboxylic acid (H2BDC), N,N-dimethylformamide (DMF) and N,N-diethylformamide (DEF) gives values of 16.69 ± 1.21 and 45.90 ± 1.46 kJ (mol Zn4O)−1, respectively. The enthalpies of interaction (ΔHint) for DMF and DEF with MOF-5 are −82.78 ± 4.84 kJ (mol DMF)−1 and −89.28 ± 3.05 kJ (mol DEF)−1, respectively. These exothermic interaction energies suggest that, at low guest loading, Lewis base solvents interact more strongly with electron accepting Zn4O clusters in the MOF than at high solvent loading. These data provide a quantitative thermodynamic basis to investigate transmetallation and solvent assisted linker exchange (SALE) methods and to synthesize new MOFs.
Co-reporter:Xiaofeng Guo, Di Wu, Hongwu Xu, Peter C. Burns, Alexandra Navrotsky
Journal of Nuclear Materials 2016 Volume 478() pp:158-163
Publication Date(Web):September 2016
DOI:10.1016/j.jnucmat.2016.06.014
The thermal decomposition of studtite (UO2)O2(H2O)2·2H2O results in a series of intermediate X-ray amorphous materials with general composition UO3+x (x = 0, 0.5, 1). As an extension of a structural study on U2O7, this work provides detailed calorimetric data on these amorphous oxygen-rich materials since their energetics and thermal stability are unknown. These were characterized in situ by thermogravimetry, and mass spectrometry. Ex situ X-ray diffraction and infrared spectroscopy characterized their chemical bonding and local structures. This detailed characterization formed the basis for obtaining formation enthalpies by high temperature oxide melt solution calorimetry. The thermodynamic data demonstrate the metastability of the amorphous UO3+x materials, and explain their irreversible and spontaneous reactions to generate oxygen and form metaschoepite. Thus, formation of studtite in the nuclear fuel cycle, followed by heat treatment, can produce metastable amorphous UO3+x materials that pose the risk of significant O2 gas. Quantitative knowledge of the energy landscape of amorphous UO3+x was provided for stability analysis and assessment of conditions for decomposition.
Hybrid perovskites, especially methylammonium lead iodide (MAPbI3), exhibit excellent solar power conversion efficiencies. However, their application is plagued by poor chemical and structural
stability. Using direct calorimetric measurement of heats of formation, MAPbI3 is shown to be thermodynamically unstable with respect to decomposition to lead iodide and methylammonium iodide, even in
the absence of ambient air or light or heat-induced defects, thus limiting its long-term use in devices. The formation enthalpy
from binary halide components becomes less favorable in the order MAPbCl3, MAPbBr3, MAPbI3, with only the chloride having a negative heat of formation. Optimizing the geometric match of constituents as measured by
the Goldschmidt tolerance factor provides a potentially quantifiable thermodynamic guide for seeking chemical substitutions
to enhance stability.
Many metal–organic frameworks (MOFs) of ultrahigh porosity (with molar volumes more than ten times greater than those of the corresponding dense phases) have been synthesized. However, the number of possible structures far exceeds those that have been made. It is logical to ask if there are energetic barriers to the stability of ultraporous MOFs or whether there is little thermodynamic penalty to their formation. Herein, we show that although the molar volumes of MOF-177 and UMCM-1 reach ultrahigh values, their energetic metastability is in the same range (of 7–36 kJ mol−1) as that seen previously for other porous materials. These findings suggest that there is little thermodynamic penalty for the synthesis of structures with varying porosity, and hence, ultraporous frameworks are energetically accessible. Therefore, innovative synthesis methods have the possibility to overcome the drawbacks of conventional approaches and greatly extend the number, porosity, and properties of new framework materials.
The Journal of Physical Chemistry C 2016 Volume 120(Issue 14) pp:7562-7567
Publication Date(Web):March 24, 2016
DOI:10.1021/acs.jpcc.5b12239
The enthalpy of water adsorption (Δh) on the metal–organic framework (MOF) HKUST-1 has been determined directly by calorimetry. The most exothermic value of Δh [−119.4 ± 0.5 kJ/(mol of water)] occurs at zero coverage and perhaps represents water confinement in the smallest (4-Å) cages. An intermediate Δh value of −50.2 ± 1.8 kJ/(mol of water) at higher loading probably corresponds to the binding of water on the available Cu nodes and subsequent filling of the largest (11-Å) pores. The weakest interactions take place in the medium (10-Å) cages, showing weak inclusion of water clusters in a limited hydrophobic environment. By combining ethanol adsorption calorimetry, mathematical analysis of the slope of the water adsorption isotherm, and the differential enthalpy of water adsorption curve, we are able not only to develop an approach to separate energetically multistage guest–host interactions in complex MOF architectures but also to distinguish a sequence of interactions with very similar energetic effects.
The Journal of Physical Chemistry C 2016 Volume 120(Issue 28) pp:15251-15256
Publication Date(Web):June 30, 2016
DOI:10.1021/acs.jpcc.6b04840
Alkali and alkaline earth ion-exchanged zeolite A samples were synthesized in aqueous exchange media. They were thoroughly studied by powder X-ray diffraction (XRD), electron microprobe (EMPA), thermogravimetric analysis and differential scanning calorimetry (TG-DSC), and high temperature oxide melt solution calorimetry. The hydration energetics and enthalpies of formation of these zeolite A materials from constituent oxides were determined. Specifically, the hydration level of zeolite A has a linear dependence on the average ionic potential (Z/r) of the cation, from 0.894 (Rb-A) to 1.317 per TO2 (Mg-A). The formation enthalpies from oxides (25 °C) range from −93.71 ± 1.77 (K-A) to −48.02 ± 1.85 kJ/mol per TO2 (Li-A) for hydrated alkali ion-exchanged zeolite A, and from −47.99 ± 1.20 (Ba-A) to −26.41 ± 1.71 kJ/mol per TO2 (Mg-A) for hydrated alkaline earth ion-exchanged zeolite A. The formation enthalpy from oxides generally becomes less exothermic as Z/r increases, but a distinct difference in slope is observed between the alkali and the alkaline earth series.
The Journal of Physical Chemistry C 2016 Volume 120(Issue 30) pp:16767-16781
Publication Date(Web):July 11, 2016
DOI:10.1021/acs.jpcc.6b04747
Bastnäsite is a fluoro-carbonate mineral that is the largest source of rare earth elements (REEs) such as Y, La, and Ce. With increasing demand for REE in many emerging technologies, there is an urgent need for improving the efficiency of ore beneficiation by froth flotation. To design improved flotation agents that can selectively bind to the mineral surface, a fundamental understanding of the bulk and surface properties of bastnäsite is essential. Unexpectedly, density functional theory (DFT) calculations using the PBEsol exchange correlation functional and the DFT-D3 dispersion correction reveal that the most stable form of La-bastnäsite is isomorphic to the structure of Ce-bastnäsite belonging to the P6̅2c space group, whereas the common structure listed in the Inorganic Crystal Structure Database structure belonging to the P6̅2m space group is ca. 11.3 kJ/mol higher in energy per LaFCO3 formula unit. We report powder X-ray diffraction measurements on synthetic La-bastnäsite to support these theoretical findings. Six different surfaces are studied by DFT, namely, [101̅0], [0001], [101̅1], [101̅2], [101̅4], and [112̅2]. Among these, the [101̅0] surface is the most stable with a surface energy of 0.73 J/m2 in vacuum and 0.45 J/m2 in aqueous solution. The shape of a La-bastnäsite nanoparticle is predicted via thermodynamic Wulff construction to be a hexagonal prism with [101̅0] and [0001] facets, chiseled at its ends by the [101̅1] and [101̅2] facets. The average surface energy of the nanoparticle in the gas phase is estimated to be 0.86 J/m2, in good agreement with a value of 1.11 J/m2 measured by calorimetry. The calculated adsorption energy of a water molecule varies widely with the surface plane and specific adsorption sites within each facet. The first layer of water molecules is predicted to adsorb strongly on the La-bastnäsite surface, in agreement with water adsorption calorimetry experiments. Our work provides an important step toward a detailed atomistic understanding of the bastnäsite–water interface and designing collector molecules that can bind specifically to bastnäsite.
The formation enthalpies from binary oxides of LiMn2O4, LiMn2−xCrxO4 (x=0.25, 0.5, 0.75 and 1), LiMn2−xFexO4 (x=0.25 and 0.5), LiMn2−xCoxO4 (x=0.25, 0.5, and 0.75) and LiMn1.75Ni0.25O4 at 25 °C were measured by high temperature oxide melt solution calorimetry and were found to be strongly exothermic. Increasing the Cr, Co, and Ni content leads to more thermodynamically stable spinels, but increasing the Fe content does not significantly affect the stability. The formation enthalpies from oxides of the fully substituted spinels, LiMnMO4 (M=Cr, Fe and Co), become more exothermic (implying increasing stability) with decreasing ionic radius of the metal and lattice parameters of the spinel. The trend in enthalpy versus metal content is roughly linear, suggesting a close-to-zero heat of mixing in LiMn2O4—LiMnMO4 solid solutions. These data confirm that transition-metal doping is beneficial for stabilizing these potential cathode materials for lithium-ion batteries.
Co-reporter:Geetu Sharma, Michael NaguibDawei Feng, Yury Gogotsi, Alexandra Navrotsky
The Journal of Physical Chemistry C 2016 Volume 120(Issue 49) pp:28131-28137
Publication Date(Web):November 19, 2016
DOI:10.1021/acs.jpcc.6b10241
MXenes are layered two-dimensional materials with exciting properties useful to a wide range of energy applications. They are derived from ceramics (MAX phases) by leaching, and their properties reflect their resulting complex compositions which include intercalating cations and anions and water. Their thermodynamic stability is likely linked to these functional groups but has not yet been addressed by quantitative experimental measurements. We report enthalpies of formation from the elements at 25 °C measured using high temperature oxide melt solution calorimetry for a layered Ti–Al–C MAX phase, and the corresponding Ti–C based MXene. The thermodynamic stability of the Ti3C2Tx MXene (Tx stands for anionic surface moieties, and intercalated cations) was assessed by calculating the enthalpy of reaction of the MAX phase (ideal composition Ti3AlC2) to form MXene. The very exothermic enthalpy of reaction confirms the stability of MXene in an aqueous environment. The surface terminations (O, OH, and F) and cations (Li) chemisorbed on the surface and intercalated in the interlayers play a major role in the thermodynamic stabilization of MXene. These findings help in understanding and potentially improving properties and performance by characterizing the energetics of species binding to MXene surfaces during synthesis and in energy storage, water desalination, and other applications.
Co-reporter:Lili Wu, Jacob Schliesser, Brian F. Woodfield, Hongwu Xu, Alexandra Navrotsky
The Journal of Chemical Thermodynamics 2016 Volume 93() pp:1-7
Publication Date(Web):February 2016
DOI:10.1016/j.jct.2015.09.019
•Heat capacities of Sr-, Rb- and Cs-hollandite were measured from T = (2 to 300) K.•Quantum Design Physical Property Measurement System (PPMS) was used.•Heat capacities were fitted to equations in three temperature ranges.•Standard entropies and Gibbs energies were calculated.•This work is relevant to nuclear waste containment using hollandite ceramics.Heat capacities of Sr-, Rb-, and Cs-hollandite with the compositions Ba1.14Sr0.10Al2.38Ti5.59O16, Ba1.17Rb0.19Al2.46Ti5.53O16, and Ba1.18Cs0.21Al2.44Ti5.53O16 were measured from T = (2 to 300) K using a Quantum Design Physical Property Measurement System (PPMS). From the heat capacity results, the following thermodynamic parameters have been determined. The characteristic Debye temperatures ΘD over the temperature range (30 to 300) K of Sr-, Rb-, and Cs-hollandite are T = (178.2, 189.7, and 189.2) K, respectively, and their standard entropies at T = 298.15 K are (413.9 ± 8.3), (415.1 ± 8.3), and (419.6 ± 8.4) J · K−1 · mol−1. Combined with previously reported formation enthalpies, their corresponding Gibbs energies of formation from oxides (ΔfGox°) are (−194.9 ± 11.4), (−195.0 ± 12.8), and (−201.1 ± 12.8) kJ · mol−1, and those from elements (ΔfGel°) are (−7694.6 ± 12.5), (−7697.0 ± 13.9), and (−7697.1 ± 13.9) kJ · mol−1 at T = 298.15 K. The similarities among the obtained ΔfGox° values suggest that the three substituted hollandites have similar thermodynamic stabilities at standard conditions, which is in agreement with the ease of Cs–Rb–Sr substitutions in the hollandite structure.
Co-reporter:G. P. Nagabhushana; Radha Shivaramaiah
Journal of the American Chemical Society 2015 Volume 137(Issue 32) pp:10351-10356
Publication Date(Web):July 27, 2015
DOI:10.1021/jacs.5b06146
Organic–inorganic hybrid materials have enormous potential for applications in catalysis, gas storage, sensors, drug delivery, and energy generation, among others. A class of hybrid materials adopts the ABX3 perovskite topology. We report here the synthesis and characterization of an isostructural series of dense hybrid perovskites, [(CH3)2NH2][M(HCOO)3], with M = Mn, Co, Ni, and Zn. These compounds have shown promising multiferroic behavior. Understanding their stability is crucial for their practical application. We report their formation enthalpies based on direct measurement by room-temperature acid solution calorimetry. The enthalpy of formation of this dimethylammonium metal formate series becomes less exothermic in the order Mn, Zn, Co, Ni. The stability of the hybrid perovskite decreases as the tolerance factor increases, unlike trends seen in inorganic perovskites. However, the trends are similar to those seen in a number of ternary transition metal oxides, suggesting that specific bonding interactions rather than geometric factors dominate the energetics.
Co-reporter:D. Wu, T. M. McDonald, Z. Quan, S. V. Ushakov, P. Zhang, J. R. Long and A. Navrotsky
Journal of Materials Chemistry A 2015 vol. 3(Issue 8) pp:4248-4254
Publication Date(Web):14 Jan 2015
DOI:10.1039/C4TA06496H
For coordinatively unsaturated metal–organic frameworks (MOFs), the metal centers can be functionalized as CO2 capture/storage adsorbents by grafting species having specific active groups. We report direct measurement of enthalpy of adsorption of CO2 on an alkylamine-appended MOF, mmen-Mg2(dobpdc) employing gas adsorption calorimetry at 298, 323 and 348 K. This methodology provides, for the first time, the detailed dependence of energy and entropy of sorption as a function of coverage and temperature. The enthalpy data suggest three types of adsorption events: strongest exothermic initial chemisorption at low coverage, majority moderate chemisorption at intermediate loading and weakest physisorption at highest coverage. The partial molar properties and isotherms are consistent with the presence of two different potential chemisorption mechanisms: 2:1 (amine–CO2) stoichiometry near zero coverage and 1:1 afterwards. Both chemical potential and differential enthalpy of adsorption become less negative with increasing temperature, implying increasing adsorbent entropy at elevated temperature. These observations are consistent with weaker CO2 binding at higher temperature.
Co-reporter:A. V. Radha, L. Lander, G. Rousse, J. M. Tarascon and A. Navrotsky
Journal of Materials Chemistry A 2015 vol. 3(Issue 6) pp:2601-2608
Publication Date(Web):17 Dec 2014
DOI:10.1039/C4TA05066E
Rational design and development of new Li-polyanion battery materials by exercising synthetic control have led to a new class of Li2M(SO4)2 compounds in monoclinic (M = Mn, Fe, Co) and orthorhombic (M = Fe, Co, Ni) polymorphic forms, using solid state (ceramic) and ball milling methods respectively. The enthalpies of formation from binary sulfates determined using isothermal acid solution calorimetry are positive, and show decrease in energetic metastability with increase in ionic radius for both monoclinic and orthorhombic (except for Ni) polymorphs. The higher symmetry orthorhombic polymorphs with Fe and Co are energetically less stable than the corresponding monoclinic polymorphs. The vibrational/rotational disorder of the SO4 tetrahedra is identified as the most likely cause of the entropy term (TΔS) of the free energy to overcome the positive enthalpy of formation in monoclinic and orthorhombic phases. Driven by thermodynamic metastability, the orthorhombic Li2M(SO4)2 (M = Fe, Co) polymorphs transform irreversibly into the monoclinic phase on heat treatment. Orthorhombic Li2Ni(SO4)2, formed by a ceramic route is thermodynamically stable and does not transform to the monoclinic phase on heating. The formation of metastable orthorhombic samples by ball milling is consistent with earlier thermodynamic studies on other Li-hydroxy/fluorosulfate systems, for which metastable tavorite polymorphs could be formed only by mild chemical synthetic approaches. This work demonstrates that the entropy term can play a key role for the synthesis, stability and phase transformation among polymorphs of Li-polyanionic compounds.
Co-reporter:Sulata Kumari Sahu, Speranta Tanasescu, Barbara Scherrer, Cornelia Marinescu and Alexandra Navrotsky
Journal of Materials Chemistry A 2015 vol. 3(Issue 38) pp:19490-19496
Publication Date(Web):21 Aug 2015
DOI:10.1039/C5TA03655K
Lanthanide cobalt perovskites LnCoO3−δ (Ln = La, Nd, Sm, and Gd) are important materials for electroceramics, catalysts, and electrodes in solid oxide fuel cells. Formation enthalpies of LnCoO3−δ compounds were measured using high temperature oxide melt solution calorimetry. The formation enthalpies of LaCoO2.992, NdCoO2.985, SmCoO2.982 and GdCoO2.968 from constituent binary oxides (Ln2O3, CoO) and O2 gas are −111.87 ± 1.36, −98.49 ± 1.33, −91.56 ± 1.46 and −88.16 ± 1.45 kJ mol−1, respectively. Thus these perovskites become energetically less stable with decrease in ionic radius of the lanthanide (from La to Gd), which corresponds to a decreasing tolerance factor and increasing oxygen deficiency. The thermodynamic stability of LaCoO2.992, NdCoO2.985, SmCoO2.982 and GdCoO2.968 was also assessed considering their oxygen partial pressures for decomposition, with good agreement between thermochemical and equilibrium data.
Laboratory synthesis of layered double hydroxides (LDH) often results in materials replete with stacking faults. Faults are known to affect several properties including sorption, electrochemical, and catalytic activity of this important class of materials. Understanding the occurrence of faults thus calls for a comprehensive analysis of formation and stability of ordered and faulted LDHs. High-temperature oxide melt solution calorimetric measurements made on an ordered and a faulted Mg–Al LDH with carbonate interlayer anion shows that ordered LDH is energetically more stable than the faulted one by ∼6 kJ/mol. The stacking faults are an intergrowth of 3R1 and 2H1 polytypes, and faults could thus mediate transformation of 3R1 to 2H1 polytypes. Several factors including pH and temperature of precipitation also affect layer stacking. The formation of stacking faults could therefore have its origin in kinetics. Water content in the interlayer also affects layer stacking, and hence it may affect properties of LDH. Improved understanding of the distribution of water molecules in LDH is also crucial in an environmental context, as LDH occur as minerals and are important for contaminant amelioration in the environment. Water adsorption calorimetry on dehydrated LDH shows a continuous decrease in the magnitude of adsorption enthalpy with increasing coverage, indicating the presence of energetically heterogeneous sites where the water molecules reside. The results also indicate that the energy of several sites where the water molecules may reside (whether in the interlayer or on the surface) overlaps, and hence it is hard to differentiate among them.
The garnet structure has been proposed as a potential crystalline nuclear waste form for accommodation of actinide elements, especially uranium (U). In this study, yttrium iron garnet (YIG) as a model garnet host was studied for the incorporation of U analogs, cerium (Ce) and thorium (Th), incorporated by a charge-coupled substitution with calcium (Ca) for yttrium (Y) in YIG, namely, 2Y3+ = Ca2+ + M4+, where M4+ = Ce4+ or Th4+. Single-phase garnets Y3–xCa0.5xM0.5xFe5O12 (x = 0.1–0.7) were synthesized by the citrate–nitrate combustion method. Ce was confirmed to be tetravalent by X-ray absorption spectroscopy and X-ray photoelectron spectroscopy. X-ray diffraction and 57Fe–Mössbauer spectroscopy indicated that M4+ and Ca2+ cations are restricted to the c site, and the local environments of both the tetrahedral and the octahedral Fe3+ are systematically affected by the extent of substitution. The charge-coupled substitution has advantages in incorporating Ce/Th and in stabilizing the substituted phases compared to a single substitution strategy. Enthalpies of formation of garnets were obtained by high temperature oxide melt solution calorimetry, and the enthalpies of substitution of Ce and Th were determined. The thermodynamic analysis demonstrates that the substituted garnets are entropically rather than energetically stabilized. This suggests that such garnets may form and persist in repositories at high temperature but might decompose near room temperature.
Co-reporter:Lei Zhang, Jonathan M. Solomon, Mark Asta, Alexandra Navrotsky
Acta Materialia 2015 Volume 97() pp:191-198
Publication Date(Web):15 September 2015
DOI:10.1016/j.actamat.2015.06.048
Abstract
The energetics of rare earth substituted UO2 solid solutions (U1−xLnxO2−0.5x+y, where Ln = La, Y, and Nd) are investigated employing a combination of calorimetric measurements and density functional theory based computations. Calculated and measured formation enthalpies agree within 10 kJ/mol for stoichiometric oxygen/metal compositions. To better understand the factors governing the stability and defect binding in rare earth substituted urania solid solutions, systematic trends in the energetics are investigated based on the present results and previous computational and experimental thermochemical studies of rare earth substituted fluorite oxides (A1−xLnxO2−0.5x, where A = Hf, Zr, Ce, and Th). A consistent trend towards increased energetic stability with larger size mismatch between the smaller host tetravalent cation and the larger rare earth trivalent cation is found for both actinide and non-actinide fluorite oxide systems where aliovalent substitution of Ln cations is compensated by oxygen vacancies. However, the large exothermic oxidation enthalpy in the UO2 based systems favors oxygen rich compositions where charge compensation occurs through the formation of uranium cations with higher oxidation states.
Co-reporter:Sulata K. Sahu, Baiyu Huang, Kristina Lilova, Brian F. Woodfield and Alexandra Navrotsky
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 34) pp:22286-22295
Publication Date(Web):22 Jul 2015
DOI:10.1039/C5CP02972D
High temperature oxide melt solution calorimetry has been performed to investigate the enthalpies of mixing (ΔmixH) of bulk and nanophase (1 − x)Fe3O4–xM3O4 (M = Co, Mn) spinel solid solutions. The entropies of mixing (ΔmixS) were calculated from the configurational entropies based on cation distributions, and the Gibbs free energies of mixing (ΔmixG) were obtained. The ΔmixH and ΔmixG for the (1 − x)Fe3O4–xCo3O4 system are negative over the complete solid solution range, for both macroscopic and nanoparticulate materials. In (1 − x)Fe3O4–xMn3O4, the formation enthalpies of cubic Fe3O4 (magnetite) and tetragonal Mn3O4 (hausmannite) are negative for Mn3O4 mole fractions less than 0.67 and slightly positive for higher manganese content. Relative to cubic Fe3O4 and cubic Mn3O4 (stable at high temperature), the enthalpies and Gibbs energies of mixing are negative over the entire composition range. A combination of measured mixing enthalpies and reported Gibbs energies in the literature provides experimental entropies of mixing. The experimental entropies of mixing are consistent with those calculated from cation distributions for x > 0.3 but are smaller than those predicted for x < 0.3. This discrepancy may be related to the calculations, having treated Fe2+ and Fe3+ as distinguishable species. The measured surface energies of the (1 − x)Fe3O4–xM3O4 solid solutions are in the range of 0.6–0.9 J m−2, similar to those of many other spinels. Because the surface energies are relatively constant, the thermodynamics of mixing at a given particle size throughout the solid solution can be considered independent of the particular particle size, thus confirming and extending the conclusions of a recent study on iron spinels.
Co-reporter:H. Sun, D. Wu, X. Guo, B. Shen and A. Navrotsky
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 17) pp:11198-11203
Publication Date(Web):25 Mar 2015
DOI:10.1039/C5CP01133G
A series of calcium-exchanged zeolite A samples with different degrees of exchange were prepared. They were characterized by powder X-ray diffraction (XRD) and differential scanning calorimetry (DSC). High temperature oxide melt drop solution calorimetry measured the formation enthalpies of hydrated zeolites CaNa-A from constituent oxides. The water content is a linear function of the degree of exchange, ranging from 20.54% for Na-A to 23.77% for 97.9% CaNa-A. The enthalpies of formation (from oxides) at 25 °C are −74.50 ± 1.21 kJ mol−1 TO2 for hydrated zeolite Na-A and −30.79 ± 1.64 kJ mol−1 TO2 for hydrated zeolite 97.9% CaNa-A. Dehydration enthalpies obtained from differential scanning calorimetry are 32.0 kJ mol−1 H2O for hydrated zeolite Na-A and 20.5 kJ mol−1 H2O for hydrated zeolite 97.9% CaNa-A. Enthalpies of formation of Ca-exchanged zeolites A are less exothermic than for zeolite Na-A. A linear relationship between the formation enthalpy and the extent of calcium substitution was observed. The energetic effect of Ca-exchange on zeolite A is discussed with an emphasis on the complex interactions between the zeolite framework, cations, and water.
Co-reporter:H. Sun, D. Wu, X. Guo and A. Navrotsky
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 14) pp:9241-9247
Publication Date(Web):26 Feb 2015
DOI:10.1039/C5CP00016E
The properties of zeolite A change significantly upon sodium–calcium exchange. The impact of cation composition on the temperature-induced phase transformations and energetics of Na–Ca exchanged zeolite A was studied systematically using powder X-ray diffraction (XRD), thermogravimetric analysis (TGA), differential scanning calorimetry (DSC) and high-temperature oxide melt solution calorimetry. As the temperature increases, the structural evolution of each Na–Ca exchanged zeolite A sample undergoes three distinct stages – dehydration, amorphization, and densification/recrystallization. Initially complete dehydration does not result in framework degradation, but further heating leads to zeolite phase degradation into other aluminosilicate phases. Both amorphization and recrystallization shift to higher temperatures as the calcium content increases. On the other hand, the enthalpies of formation for the high temperature aluminosilicate phases, the amorphous phase (AP) and the dense phase (DP), appear to be a linear function of calcium content (average ionic potential) with diminishing of energetic stability upon increasing the Ca content. 100% Na-A heated at 1200 °C has the most exothermic enthalpy of formation from oxides (−65.87 ± 0.87 kJ mol−1 – TO2), while 97.9% CaNa-A heated at 945 °C has the least exothermic value (−5.26 ± 0.62 kJ mol−1 – TO2). For different aluminosilicates with the same chemical composition, the dense phase (DP) assemblage is more stable than the amorphous phase (AP).
Interaction of carbonate surfaces with water plays a crucial role in carbonate nucleation and crystal growth. This study provides experimental evidence for the existence of two different types of water having distinct energetics in amorphous carbonates, MCO3 (M = Ca, Mn, and Mg). The adsorption enthalpy curves obtained using a combination of gas sorption and microcalorimetry show two different energetic regions, which correspond to weakly bound restrictedly mobile and strongly bound rigid H2O components. For weakly bound water, adsorption enthalpies of amorphous calcium carbonate (ACC) (−55.3 ± 0.9 kJ/mol), amorphous manganese carbonate (AMnC) (−54.1 ± 0.8 kJ/mol), and amorphous magnesium carbonate (AMgC) (−56.1 ± 0.4 kJ/mol) fall in the same range, suggesting their interaction modes may be similar in all amorphous phases. Water adsorption enthalpies of crystalline nanocalcite (−96.3 ± 1 kJ/mol) and nano-MnCO3 (−65.3 ± 3 kJ/mol) measured in previous studies are more exothermic than values for ACC (−62.1 ± 0.7 kJ/mol) and AMnC (−54.1 ± 0.8 kJ/mol) and could provide a driving force for crystallization of ACC and AMnC in the presence of water. The differences in water adsorption behavior between amorphous and naocrystalline material have significant implications for crystal growth, biomineralization, and carbonate geochemistry.
Co-reporter:A. Mielewczyk-Gryn, S. Wachowski, K.I. Lilova, X. Guo, M. Gazda, A. Navrotsky
Ceramics International 2015 Volume 41(Issue 2) pp:2128-2133
Publication Date(Web):March 2015
DOI:10.1016/j.ceramint.2014.10.010
Analysis of the influence of antimony substitution on the temperature dependence of unit cell distortion in lanthanum orthoniobate has been performed. The values of spontaneous strain and Landau order parameter for three different antimony contents have been calculated. The monoclinic–tetragonal phase transition occurring for antimony substituted lanthanum orthoniobate was found to be of the second order. High temperature oxide melt solution calorimetry has shown that antimony substitution has little influence on the stability of the monoclinic phase. The average value of enthalpy of formation of antimony substituted lanthanum orthoniobate with fergusonite structure is −132.0±0.8 kJ/mol, and that of scheelite structure is −126.4±1.5 kJ/mol, implying greater stability for the former polymorph.
Journal of Nuclear Materials 2015 Volume 465() pp:682-691
Publication Date(Web):October 2015
DOI:10.1016/j.jnucmat.2015.06.059
•We synthesize, characterize LnxU1−xO2−0.5x+y solid solutions (Ln = La, Y, Nd).•Formation enthalpies become more exothermic with increasing rare earth content.•Oxidation enthalpy of LnxU1−xO2−0.5x+y is similar to that of UO2 to UO3.•Direct calorimetric measurements are in good agreement with free energy data.Lanthanum, yttrium, and neodymium doped uranium dioxide samples in the fluorite structure have been synthesized, characterized in terms of metal ratio and oxygen content, and their enthalpies of formation measured by high temperature oxide melt solution calorimetry. For oxides doped with 10–50 mol % rare earth (Ln) cations, the formation enthalpies from constituent oxides (LnO1.5, UO2 and UO3 in a reaction not involving oxidation or reduction) become increasingly exothermic with increasing rare earth content, while showing no significant dependence on the varying uranium oxidation state. The oxidation enthalpy of LnxU1−xO2−0.5x+y is similar to that of UO2 to UO3 for all three rare earth doped systems. Though this may suggest that the oxidized uranium in these systems is energetically similar to that in the hexavalent state, thermochemical data alone can not constrain whether the uranium is present as U5+, U6+, or a mixture of oxidation states. The formation enthalpies from elements calculated from the calorimetric data are generally consistent with those from free energy measurements.
Co-reporter:Dana Reusser, Jacob Schliesser, Brian F. Woodfield, Alexandra Navrotsky
The Journal of Chemical Thermodynamics 2015 Volume 89() pp:296-305
Publication Date(Web):October 2015
DOI:10.1016/j.jct.2015.06.001
•Calorimetry was used to measure ΔdsHΔdsH for a germanium – substituted Al13 selenate.•The enthalpies, ΔfH°ΔfH° and Δf,oxHΔf,oxH, were determined for this GeAl12 selenate.•Heat capacities were measured on the MAl12 selenates, M = Al(III), Ga(III) or Ge(IV).•Standard entropies and free energies were determined for these MAl12 selenates.•Energetics of AlAl12 and GaAl12 were similar; GeAl12 was found to be less stable.We report enthalpies of formation from the elements and oxides, ΔfH°ΔfH° = −(22,673.44 ± 30.19) kJ·mol-1kJ·mol-1 and Δf,oxHΔf,oxH = −(869.68 ± 28.75) kJ·mol-1kJ·mol-1, for [GeO4Al12(OH)24(H2O)12](SeO4)4·12H2O(cr) (GeAl12) measured using high temperature oxide-melt solution calorimetry. This material is the selenate salt of the germanium-substituted polynuclear Al137+ ion in the ε-Keggin structure. We also report heat capacities from temperatures of (2 to 300) K on this GeAl12 selenate and its other heterometal substituted forms, Na[AlO4Al12(OH)24(H2O)12](SeO4)4·12H2O(cr) (AlAl12) and Na[GaO4Al12(OH)24(H2O)12](SeO4)4·12H2O(cr) (GaAl12), measured using a Quantum Design Physical Property Measurement System (PPMS). These measurements were used to calculate entropies and subsequently free energies of formation for these three materials. All three MAl12 selenates, where M = Al(III), Ga(III), or Ge(IV), have similar heat capacities from temperatures (2 to 300) K and similar characteristic Debye temperatures, ΘD, suggesting similar lattice vibrational densities of states.
Co-reporter:Dr. Dana Reusser; William H. Casey; Alexra Navrotsky
Angewandte Chemie International Edition 2015 Volume 54( Issue 32) pp:9253-9256
Publication Date(Web):
DOI:10.1002/anie.201503544
Abstract
The ε-Keggin [AlO4Al12(OH)24(H2O)12]7+ ion (AlAl127+) is a metastable precursor in the formation of aluminum oxyhydroxide solids. It also serves as a useful model for the chemistry of aluminous mineral surfaces. Herein we calculate the enthalpies of formation for this aqueous ion and its heterometal-substituted forms, GaAl127+ and GeAl128+, using solution calorimetry. Rather than measuring the enthalpies of the MAl127/8+ ions directly from solution hydrolysis, we measured the metathesis reaction of the crystallized forms with barium chloride creating an aqueous aluminum solution monospecific in MAl127/8+. Then, the contributions to the heat of formation from the crystallized forms were subtracted using referenced states. When comparing the aqueous AlAl127+ ion to solid aluminum (oxy)-hydroxide phases, we found that this ion lies closer in energy to solid phases than to aqueous aluminum monomers, thus explaining its role as a precursor to amorphous aluminum hydroxide phases.
The ε-Keggin [AlO4Al12(OH)24(H2O)12]7+ ion (AlAl127+) is a metastable precursor in the formation of aluminum oxyhydroxide solids. It also serves as a useful model for the chemistry of aluminous mineral surfaces. Herein we calculate the enthalpies of formation for this aqueous ion and its heterometal-substituted forms, GaAl127+ and GeAl128+, using solution calorimetry. Rather than measuring the enthalpies of the MAl127/8+ ions directly from solution hydrolysis, we measured the metathesis reaction of the crystallized forms with barium chloride creating an aqueous aluminum solution monospecific in MAl127/8+. Then, the contributions to the heat of formation from the crystallized forms were subtracted using referenced states. When comparing the aqueous AlAl127+ ion to solid aluminum (oxy)-hydroxide phases, we found that this ion lies closer in energy to solid phases than to aqueous aluminum monomers, thus explaining its role as a precursor to amorphous aluminum hydroxide phases.
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 13) pp:2439-2443
Publication Date(Web):June 9, 2015
DOI:10.1021/acs.jpclett.5b00893
Metal–organic frameworks (MOFs) can be engineered as natural gas storage materials by tuning the pore structures and surface properties. Here we report the direct measurement of CH4 adsorption enthalpy on a paddlewheel MOF (Cu HKUST-1) using gas adsorption calorimetry at 25 °C at low pressures (below 1 bar). In this pressure region, the CH4–CH4 intermolecular interactions are minimized and the energetics solely reflects the CH4–MOF interactions. Our results suggest moderately exothermic physisorption with an enthalpy of −21.1 ± 1.1 kJ/mol CH4 independent of coverage. This calorimetric investigation complements previous computational and crystallographic studies by providing zero coverage enthalpies of CH4 adsorption. The analysis of the new and literature data suggests that in initial stages of adsorption the CH4–HKUST-1 interaction tends to be more sensitive to the pore dimension than to the guest polarizability, suggesting a less specific chemical binding role for the open Cu site.
Co-reporter:Alexandra Navrotsky, Wingyee Lee, Aleksandra Mielewczyk-Gryn, Sergey V. Ushakov, Andre Anderko, Haohan Wu, Richard E. Riman
The Journal of Chemical Thermodynamics 2015 Volume 88() pp:126-141
Publication Date(Web):September 2015
DOI:10.1016/j.jct.2015.04.008
•Rare earths are used in applications involving multicomponent oxide materials.•This paper summarizes thermodynamic properties of these diverse oxide materials.•Ionic size is a major factor in determining the systematics of phase stabilityRare earth elements (RE) are incorporated into a large variety of complex oxide phases to provide tailored mechanical, electrical, optical, and magnetic properties. Thermodynamics control phase stability, materials compatibility in use, corrosion, and transformation. This review presents, in one compilation, the thermodynamic properties of a large number of such materials and discusses systematic trends in energetics and the factors controlling stability.Graphical abstract
Knowing the nature of interactions between small organic molecules and surfaces of nanoparticles (NP) is crucial for fundamental
understanding of natural phenomena and engineering processes. Herein, we report direct adsorption enthalpy measurement of
ethanol on a series of calcite nanocrystals, with the aim of mimicking organic–NP interactions in various environments. The
energetics suggests a spectrum of adsorption events as a function of coverage: strongest initial chemisorption on active sites
on fresh calcite surfaces, followed by major chemical binding to form an ethanol monolayer and, subsequently, very weak, near-zero
energy, physisorption. These thermochemical observations directly support a structure where the ethanol monolayer is bonded
to the calcite surface through its polar hydroxyl group, leaving the hydrophobic ends of the ethanol molecules to interact
only weakly with the next layer of adsorbing ethanol and resulting in a spatial gap with low ethanol density between the monolayer
and subsequent added ethanol molecules, as predicted by molecular dynamics and density functional calculations. Such an ordered
assembly of ethanol on calcite NP is analogous to, although less strongly bonded than, a capping layer of organics intentionally
introduced during NP synthesis, and suggests a continuous variation of surface structure depending on molecular chemistry,
ranging from largely disordered surface layers to ordered layers that nevertheless are mobile and can rearrange or be displaced
by other molecules to strongly bonded immobile organic capping layers. These differences in surface structure will affect
chemical reactions, including the further nucleation and growth of nanocrystals on organic ligand-capped surfaces.
Co-reporter:Xiaofeng Guo;Stéphanie Szenknect;Adel Mesbah;Sabrina Labs;Nicolas Clavier;Christophe Poinssot;Sergey V. Ushakov;Hildegard Curtius;Dirk Bosbach;Rodney C. Ewing;Peter C. Burns;Nicolas Dacheux
PNAS 2015 112 (21 ) pp:6551-6555
Publication Date(Web):2015-05-26
DOI:10.1073/pnas.1507441112
Coffinite, USiO4, is an important U(IV) mineral, but its thermodynamic properties are not well-constrained. In this work, two different coffinite
samples were synthesized under hydrothermal conditions and purified from a mixture of products. The enthalpy of formation
was obtained by high-temperature oxide melt solution calorimetry. Coffinite is energetically metastable with respect to a
mixture of UO2 (uraninite) and SiO2 (quartz) by 25.6 ± 3.9 kJ/mol. Its standard enthalpy of formation from the elements at 25 °C is −1,970.0 ± 4.2 kJ/mol. Decomposition
of the two samples was characterized by X-ray diffraction and by thermogravimetry and differential scanning calorimetry coupled
with mass spectrometric analysis of evolved gases. Coffinite slowly decomposes to U3O8 and SiO2 starting around 450 °C in air and thus has poor thermal stability in the ambient environment. The energetic metastability
explains why coffinite cannot be synthesized directly from uraninite and quartz but can be made by low-temperature precipitation
in aqueous and hydrothermal environments. These thermochemical constraints are in accord with observations of the occurrence
of coffinite in nature and are relevant to spent nuclear fuel corrosion.
Co-reporter:Xiaofeng Guo, Amir H. Tavakoli, Steve Sutton, Ravi K. Kukkadapu, Liang Qi, Antonio Lanzirotti, Matt Newville, Mark Asta, and Alexandra Navrotsky
Chemistry of Materials 2014 Volume 26(Issue 2) pp:1133
Publication Date(Web):December 27, 2013
DOI:10.1021/cm403444f
The garnet structure is a promising nuclear waste form because it can accommodate various actinide elements. Yttrium iron garnet, Y3Fe5O12 (YIG), is a model composition for such substitutions. Since cerium (Ce) can be considered an analogue of actinide elements such as thorium (Th), plutonium (Pu), and uranium (U), studying the local structure and thermodynamic stability of Ce-substituted YIG (Ce:YIG) can provide insights into the structural and energetic aspects of large ion substitution in garnets. Single phases of YIG with Ce substitution up to 20 mol % (Y3–xCexFe5O12 with 0 ≤ x ≤ 0.2) were synthesized through a citrate–nitrate combustion method. The oxidation state of Ce was examined by X-ray absorption near edge structure spectroscopy (XANES); the oxidation state and site occupancy of iron (Fe) as a function of Ce loading also was monitored by 57Fe–Mössbauer spectroscopy. These measurements establish that Ce is predominantly in the trivalent state at low substitution levels, while a mixture of trivalent and tetravalent states is observed at higher concentrations. Fe was predominately trivalent and exists in multiple environments. High temperature oxide melt solution calorimetry was used to determine the enthalpy of formation of these Ce-substituted YIGs. The thermodynamic analysis demonstrated that, although there is an entropic driving force for the substitution of Ce for Y, the substitution reaction is enthalpically unfavorable. The experimental results are complemented by electronic structure calculations performed within the framework of density functional theory (DFT) with Hubbard-U corrections, which reproduce the observed increase in the tendency for tetravalent Ce to be present with a higher loading of Ce. The DFT+U results suggest that the energetics underlying the formation of tetravalent Ce involve a competition between an unfavorable energy to oxidize Ce and reduce Fe and a favorable contribution due to strain-energy reduction. The structural and thermodynamic findings suggest a strategy to design thermodynamically favorable substitutions of actinides in the garnet system.Keywords: calorimetry; cerium; density functional theory; Mössbauer spectroscopy; nuclear waste form; X-ray absorption spectroscopy; yttrium iron garnet;
Co-reporter:Julia V. Zaikina, Elayaraja Muthuswamy, Kristina I. Lilova, Zachary M. Gibbs, Michael Zeilinger, G. Jeffrey Snyder, Thomas F. Fässler, Alexandra Navrotsky, and Susan M. Kauzlarich
Chemistry of Materials 2014 Volume 26(Issue 10) pp:3263
Publication Date(Web):April 21, 2014
DOI:10.1021/cm5010467
A thermochemical study of three germanium allotropes by differential scanning calorimetry (DSC) and oxidative high-temperature drop solution calorimetry with sodium molybdate as the solvent is described. Two allotropes, microcrystalline allo-Ge (m-allo-Ge) and 4H-Ge, have been prepared by topotactic deintercalation of Li7Ge12 with methanol (m-allo-Ge) and subsequent annealing at 250 °C (4H-Ge). Transition enthalpies determined by differential scanning calorimetry amount to 4.96(5) ± 0.59 kJ/mol (m-allo-Ge) and 1.46 ± 0.55 kJ/mol (4H-Ge). From high-temperature drop solution calorimetry, they are energetically less stable by 2.71 ± 2.79 kJ/mol (m-allo-Ge) and 5.76 ± 5.12 kJ/mol (4H-Ge) than α-Ge, which is the stable form of germanium under ambient conditions. These data are in agreement with DSC, as well as with the previous quantum chemical calculations. The morphology of the m-allo-Ge and 4H-Ge crystallites was investigated by a combination of scanning electron microscopy, transmission electron microscopy, and atomic force microscopy. Even though the crystal structures of m-allo-Ge and 4H-Ge cannot be considered as truly layered, these phases retain the crystalline morphology of the layered precursor Li7Ge12. Investigation by diffuse reflectance infrared Fourier transform spectroscopy and UV–vis diffuse reflectance measurements reveal band gaps in agreement with quantum chemical calculations.
Co-reporter:A. V. Radha, C. V. Subban, M. L. Sun, J. M. Tarascon and A. Navrotsky
Journal of Materials Chemistry A 2014 vol. 2(Issue 19) pp:6887-6894
Publication Date(Web):27 Mar 2014
DOI:10.1039/C3TA15457B
The thermodynamic stabilities of lithium hydroxysulfates of general formula LiMSO4OH (M = Co, Fe, Mn) with layered and tavorite structures have been investigated using isothermal acid solution calorimetry. These compounds have been explored as sustainable F-free alternatives to F-based flurosulfate cathode materials. The energetic trends for layered LiMSO4OH (M = Co, Fe and Mn) samples generally showed a decrease in stability with an increase in ionic radius (Co2+ to Mn2+), reflecting weaker M–O bonds and increasing structural distortions. The low symmetry tavorite LiFeSO4OH with a structure containing corner-shared octahedral chains is less stable than layered LiFeSO4OH with a more symmetric edge-shared octahedral structure. Structural distortions within the metal octahedra as well as changes in sulfate bonding and symmetry of the SO42− groups appear to control the thermodynamic and electrochemical behavior of LiMSO4OH (M = Co, Fe and Mn) materials. Both redox potential and thermodynamic stability of layered LiMSO4OH (M = Co, Fe and Mn) can be correlated to the lowering of the sulfate bonding symmetry in the structure from C3v to C2v.
Co-reporter:M. D. Gonçalves, P. S. Maram, R. Muccillo and A. Navrotsky
Journal of Materials Chemistry A 2014 vol. 2(Issue 42) pp:17840-17847
Publication Date(Web):05 Sep 2014
DOI:10.1039/C4TA03487B
The enthalpies of formation from binary oxide components at 25 °C of Ba(Zr1−xYx)O3−δ, x = 0.1 to 0.5 solid solutions are measured by high temperature oxide melt solution calorimetry in a molten solvent, 3Na2O·4MoO3 at 702 °C. The enthalpy of formation is exothermic for all the compositions and becomes less negative when increasing yttrium content from undoped (−115.12 ± 3.69 kJ mol−1) to x = 0.5 (−77.09 ± 4.31 kJ mol−1). The endothermic contribution to the enthalpy of formation with doping content can be attributed to lattice distortions related to the large ionic radius difference of yttrium and zirconium and vacancy formation. For 0.3 ≤ x ≤ 0.5, the enthalpy of formation appears to level off, consistent with an exothermic contribution from defect clustering. Raman spectra indicate changes in short range structural features as a function of dopant content and, suggests that from x = 0.3 to 0.5 the defects begins to cluster significantly in the solid solution, which corroborates with the thermodynamic data and the drop-off in proton conductivity from x > 0.3.
Co-reporter:X. Guo, Zs. Rak, A. H. Tavakoli, U. Becker, R. C. Ewing and A. Navrotsky
Journal of Materials Chemistry A 2014 vol. 2(Issue 40) pp:16945-16954
Publication Date(Web):14 Aug 2014
DOI:10.1039/C4TA03683B
The thermodynamic stability of Th-doped yttrium iron garnet (Y3Fe5O12, YIG) as a possible actinide-bearing material has been investigated using calorimetric measurements and first-principles electronic-structure calculations. Yttrium iron garnet with thorium substitution ranging from 0.04 to 0.07 atoms per formula unit (Y3−xThxFe5O12, x = 0.04–0.07) was synthesized using a citrate–nitrate combustion method. High-temperature oxide melt solution calorimetry was used to determine their enthalpy of formation. The thermodynamic analysis demonstrates that, although the substitution enthalpy is slightly endothermic, an entropic driving force for the substitution of Th for Y leads to a near-zero change in the Gibbs free energy. First-principles calculations within the density functional theory (DFT) indicate that the main limiting factors for Th incorporation into the YIG structure are the narrow stability domain of the host YIG and the formation of ThO2 as a secondary phase. Nevertheless, the defect formation energy calculations suggest that by carefully tuning the atomic and electronic chemical potentials, Th can be incorporated into YIG. The thermodynamic results, as a whole, support the possible use of garnet phases as nuclear waste forms; however, this will require careful consideration of the repository conditions.
Co-reporter:Gustavo C.C. Costa, John K. McDonough, Yury Gogotsi, Alexandra Navrotsky
Carbon 2014 Volume 69() pp:490-494
Publication Date(Web):April 2014
DOI:10.1016/j.carbon.2013.12.053
The standard enthalpies of formation at 25 °C of onion-like carbons (OLC) with different structural ordering have been investigated by high-temperature oxidation calorimetry. In terms of enthalpy and depending on the degree of structural ordering, OLC can be up to 16 kJ mol−1 less stable than graphite but up to 27 kJ mol−1 more stable than their fullerene allotropes. Furthermore, OLC are approximately 5–9 kJ mol−1 less stable than single-wall carbon nanotubes (SWCNTs). The samples prepared at 1800 °C are energetically less stable than samples made at 1300 and 1500 °C. These changes in energetics may stem from oxygen-containing functional groups bonded to the structure or from the creation of topological defects (polygonization and pentagon formation) whose concentration increases with increasing temperature and whose higher energy is balanced by configurational entropy.
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 6) pp:2331-2337
Publication Date(Web):11 Dec 2013
DOI:10.1039/C3CP54553A
Rare-earth stabilized bismuth oxides are known for their excellent ionic conductivity at intermediate temperatures. However, previous studies have shown that their conductivity deteriorates during extended heat treatments at 500–600 °C, although the fluorite phase is maintained. In this study, the enthalpies of formation of quenched and aged ytterbia- and dysprosia-stabilized bismuth oxides were measured using high-temperature oxide melt solution calorimetry in 3Na2O–4MoO3 solvent at 702 °C. While a modest energy difference (−2 to −3 kJ mol−1) drives the kinetically slow aging transformation in the ytterbia-stabilized system at moderate dopant contents, no energetic driving force is detectable in the dysprosia-stabilized system. Although the small magnitude of the exothermic ordering energy suggests extensive short range ordering in both the quenched and aged samples, the anion configuration specific to the aged samples is nevertheless responsible for the significant decrease in conductivity.
Co-reporter:L. Wu, J. Hughes, M. Moliner, A. Navrotsky, A. Corma
Microporous and Mesoporous Materials 2014 Volume 187() pp:77-81
Publication Date(Web):15 March 2014
DOI:10.1016/j.micromeso.2013.12.013
•The thermodynamic data of two large pore pure silica BECs were provided.•The thermodynamic data of extra-large pore zeolite ITQ-33 was provided.•The energetics of two BECs relative to its structural defect was demonstrated.•The effects of Ge content, molar volume on ΔHf,ox of ITQs were studied.•The content of D4R units also effect on the stability of ITQs.The enthalpies of formation of the large pore pure silica beta polymorph C (BEC) and the extra-large pore germanosilicate ITQ-33 zeolite are investigated by high temperature oxide melt solution calorimetry. The enthalpies of formation from quartz for two BECs synthesized with different organic structure directing agents, SDA1 and SDA9 differ by 4 kJ/mol. The two SDAs produce phases with different properties as well as different energetics for the same framework and composition, due to the different amount of structural defects, while the more defective BEC is energetically less stable by 4 kJ/mol. The enthalpy of formation of defect-free pure silica BEC agrees with the predicted value proposed several years ago. Moreover, the enthalpy of formation of ITQ-33 (Ge/(Ge + Si) = 0.3) supports the energetic trends seen previously, namely that the enthalpy of formation becomes more endothermic as the content of double four rings (D4R) increases. The previous trend of energetics of porous materials versus molar volume is supported by the present data, with a diminishing destabilization for very open structures.
The Journal of Physical Chemistry C 2014 Volume 118(Issue 51) pp:29836-29844
Publication Date(Web):November 20, 2014
DOI:10.1021/jp508678k
Energetics of CO2 uptake by layered double hydroxides (LDH) of Mg and Al and their corresponding mixed metal oxides (MMO) of two different compositions (Mg/Al = 2:1 and 3:1) were investigated by gas adsorption calorimetry. The initial adsorption enthalpies for all the LDH and MMO are similar and in the range −90 to −120 kJ/mol, indicating strong chemisorption of CO2. However, the differential adsorption enthalpy varies substantially with increasing coverage for different samples. LDH prepared by coprecipitation (both Mg/Al = 2 and 3) and their corresponding MMO exhibit similar CO2 uptake, which are in the working sorption capacity range (0.6–1 mmol/g), but the one prepared by urea hydrolysis shows poor sorption capacity (0.02–0.35 mmol/g). An integral enthalpy of adsorption of −54 kJ/mol was observed for [Mg–Al–CO3] LDH prepared by urea hydrolysis and −59 kJ/mol and −57 kJ/mol for the ones prepared by coprecipitation with composition 3:1 and 2:1, respectively. Attenuated total reflectance spectroscopy measurements of samples after CO2 uptake aid in identifying the mode of binding of adsorbed carbonate, which gives information about strength of basic sites. The presence of monodentate carbonate in all samples suggests that strong basic sites are available in LDH and MMO, in agreement with the large exothermic initial adsorption enthalpies observed, indicating strong chemisorption.
The energetics of nanosized Fe/Ti spinel oxides was studied using high-temperature oxide melt solution calorimetry. The mixing properties of the solid solution in the system were obtained, and through comparison to macroscopic materials the effect of particle size on the thermodynamics was assessed. The surface energies of the nanosized materials are similar within the errors regardless of the composition, and are consistent with those determined for other spinels. The enthalpies of oxidation to hematite plus rutile of the iron titanium spinels follow a linear trend with the Fe2+ content, which allows them to be calculated for any composition or particle size. The heat of formation of the macroscopic and nanosized titanomagnetites was fit as a polynomial function and the numerical coefficients are presented. The enthalpies of mixing in the titanomagnetite and titanomaghemite solid solutions are similar at the macroscopic and nanoscale.
Co-reporter:Dr. Eitan Tiferet;Dr. Adrià Gil;Dr. Carles Bo;Dr. Tatiana Y. Shvareva;Dr. May Nyman;Dr. Alexra Navrotsky
Chemistry - A European Journal 2014 Volume 20( Issue 13) pp:3646-3651
Publication Date(Web):
DOI:10.1002/chem.201304076
Abstract
Nanoscale uranyl peroxide clusters containing UO22+ groups bonded through peroxide bridges to form polynuclear molecular species (polyoxometalates) exist both in solution and in the solid state. There is an extensive family of clusters containing 28 uranium atoms (U28 clusters), with an encapsulated anion in the center, for example, [UO2(O2)3−x(OH)x4−], [Nb(O2)43−], or [Ta(O2)43−]. The negative charge of these clusters is balanced by alkali ions, both encapsulated, and located exterior to the cluster. The present study reports measurement of enthalpy of formation for two such U28 compounds, one of which is uranyl centered and the other is peroxotantalate centered. The [(Ta(O2)4]-centered U28 capsule is energetically more stable than the [(UO2)(O2)3]-centered capsule. These data, along with our prior studies on other uranyl–peroxide solids, are used to explore the energy landscape and define thermochemical trends in alkali–uranyl–peroxide systems. It was suggested that the energetic role of charge-balancing alkali ions and their electrostatic interactions with the negatively charged uranyl–peroxide species is the dominant factor in defining energetic stability. These experimental data were supported by DFT calculations, which agree that the [(Ta(O2)4]-centered U28 capsule is more stable than the uranyl-centered capsule. Moreover, the relative stability is controlled by the interactions of the encapsulated alkalis with the encapsulated anion. Thus, the role of alkali-anion interactions was shown to be important at all length scales of uranyl–peroxide species: in both comparing clusters to clusters; and clusters to monomers or extended solids.
Chemistry - A European Journal 2014 Volume 20( Issue 13) pp:
Publication Date(Web):
DOI:10.1002/chem.201400426
Abstract
Invited for the cover of this issue are the groups of Alex Navrotsky at University of California, Davis, Carles Bo at Institute of Chemical Research of Catalonia (ICIQ) and Departament de Química Física i Inorgànica, Universitat Rovira i Virgili, and May Nyman at Oregon State University. The image depicts the stability of uranyl polyoxometalates changes as a function of the encapsulated species, which was quantified by experiment and theory. Read the full text of the article at 10.1002/chem.201304076.
Molecular-level interactions at organic–inorganic interfaces play crucial roles in many fields including catalysis, drug delivery,
and geological mineral precipitation in the presence of organic matter. To seek insights into organic–inorganic interactions
in porous framework materials, we investigated the phase evolution and energetics of confinement of a rigid organic guest,
N,N,N-trimethyl-1-adamantammonium iodide (TMAAI), in inorganic porous silica frameworks (SSZ-24, MCM-41, and SBA-15) as a function
of pore size (0.8 nm to 20.0 nm). We used hydrofluoric acid solution calorimetry to obtain the enthalpies of interaction between
silica framework materials and TMAAI, and the values range from −56 to −177 kJ per mole of TMAAI. The phase evolution as a
function of pore size was investigated by X-ray diffraction, IR, thermogravimetric differential scanning calorimetry, and
solid-state NMR. The results suggest the existence of three types of inclusion depending on the pore size of the framework:
single-molecule confinement in a small pore, multiple-molecule confinement/adsorption of an amorphous and possibly mobile
assemblage of molecules near the pore walls, and nanocrystal confinement in the pore interior. These changes in structure
probably represent equilibrium and minimize the free energy of the system for each pore size, as indicated by trends in the
enthalpy of interaction and differential scanning calorimetry profiles, as well as the reversible changes in structure and
mobility seen by variable temperature NMR.
Co-reporter:Hui Sun ; Di Wu ; Xiaofeng Guo ; Benxian Shen ; Jichang Liu
The Journal of Physical Chemistry C 2014 Volume 118(Issue 44) pp:25590-25596
Publication Date(Web):October 14, 2014
DOI:10.1021/jp508514e
Study of the energetics of confinement of small organic molecules in microporous frameworks provides essential information for rational design and application of functional porous materials. Using immersion calorimetry and temperature-programmed desorption coupled with mass spectroscopy, we describe the complex guest–host interactions of a nonpolar organic molecule (n-hexane) with a series of calcium-exchanged zeolite A materials whose pore accessibility was modified by varying the calcium content to achieve inaccessible, partially accessible, and fully accessible central cavities for n-hexane molecules. The overall magnitude and trend with the pore size of n-hexane–zeolite A interactions have been determined. In addition, the contributions to energetics from the external surface (ΔHwet), tetrahedral framework (ΔHg-h), and adjacent n-hexane molecules (ΔHg-g) were separated, quantified, and interpreted.
Co-reporter:Laurens D. A. Siebbeles;Zhongwu Wang;Hongwu Xu;Wiel H. Evers;James M. Boncella;Zewei Quan;Di Wu;Jinlong Zhu
PNAS 2014 Volume 111 (Issue 25 ) pp:9054-9057
Publication Date(Web):2014-06-24
DOI:10.1073/pnas.1408835111
Self-assembly of nanocrystals (NCs) into superlattices is an intriguing multiscale phenomenon that may lead to materials with
novel collective properties, in addition to the unique properties of individual NCs compared with their bulk counterparts.
By using different dispersion solvents, we synthesized three types of PbSe NC superlattices—body-centered cubic (bcc), body-centered tetragonal (bct), and face-centered cubic (fcc)—as confirmed by synchrotron small-angle X-ray scattering. Solution calorimetric measurements in hexane show that the enthalpy
of formation of the superlattice from dispersed NCs is on the order of −2 kJ/mol. The calorimetric measurements reveal that
the bcc superlattice is the energetically most stable polymorph, with the bct being 0.32 and the fcc 0.55 kJ/mol higher in enthalpy. This stability sequence is consistent with the decreased packing efficiency of PbSe NCs from
bcc (17.2%) to bct (16.0%) and to fcc (15.2%). The small enthalpy differences among the three polymorphs confirm a closely spaced energy landscape and explain
the ease of formation of different NC superlattices at slightly different synthesis conditions.
Angewandte Chemie International Edition 2014 Volume 53( Issue 36) pp:9517-9521
Publication Date(Web):
DOI:10.1002/anie.201404618
Abstract
Earlier studies have shown a strong correlation between the enthalpy of formation, ΔHf,ox, and the ionic conductivity, σi, near room temperature in doped ceria systems, which are promising solid electrolytes for intermediate-temperature solid oxide fuel cells (IT-SOFCs). The present work demonstrates that this correlation holds at the operating temperature of IT-SOFCs, 600–700 °C. Solid solutions of Ce1−xNdxO2−0.5x, Ce1−xSmxO2−0.5x, and Ce1−xSm0.5xNd0.5xO2−0.5x are studied. The ΔHf,ox at 702 °C is determined by considering the excess heat content between 25 and 702 °C combined with the value of ΔHf,ox at 25 °C. Both σi and ΔHf,ox show maxima at x=0.15 and 0.20 for the singly and doubly doped ceria, respectively, suggesting that the number of mobile oxygen vacancies in these solid solutions reaches a maximum near those compositions. An increase in temperature results in a shift of the maximum in both ΔHf,ox and σi towards higher concentrations. This shift results from a gradual increase in dissociation of the defect associates.
Co-reporter:Douglas Gouvêa, Sergey V. Ushakov, and Alexandra Navrotsky
Langmuir 2014 Volume 30(Issue 30) pp:9091-9097
Publication Date(Web):2017-2-22
DOI:10.1021/la500743u
Adsorption of H2O and CO2 on zinc oxide surfaces was studied by gas adsorption calorimetry on nanocrystalline samples prepared by laser evaporation in oxygen to minimize surface impurities and degassed at 450 °C. Differential enthalpies of H2O and CO2 chemisorption are in the range −150 ±10 kJ/mol and −110 ±10 kJ/mol up to a coverage of 2 molecules per nm2. Integral enthalpy of chemisorption for H2O is −96.8 ±2.5 kJ/mol at 5.6 H2O/nm2 when enthalpy of water condensation is reached, and for CO2 is −96.6 ±2.5 kJ/mol at 2.6 CO2/nm2 when adsorption ceases. These values are consistent with those reported for ZnO prepared by other methods after similar degas conditions. The similar energetics suggests possible competition of CO2 and H2O for binding to ZnO surfaces. Exposure of bulk and nanocrystalline ZnO with preadsorbed CO2 to water vapor results in partial displacement of CO2 by H2O. In contrast, temperature-programmed desorption (TPD) indicates that a small fraction of CO2 is retained on ZnO surfaces up to 800 °C, under conditions where all H2O is desorbed, with adsorption energies near −200 kJ/mol. Although molecular mechanisms of adsorption were not studied, the thermodynamic data are consistent with dissociative adsorption of H2O at low coverage and with several different modes of CO2 binding.
Co-reporter:Sergey V. Ushakov;Xiaofeng Guo;Sabrina Labs;Dirk Bosbach;Hildegard Curtius
PNAS 2014 Volume 111 (Issue 50 ) pp:17737-17742
Publication Date(Web):2014-12-16
DOI:10.1073/pnas.1421144111
Metastudtite, (UO2)O2(H2O)2, is one of two known natural peroxide minerals, but little is established about its thermodynamic stability. In this work,
its standard enthalpy of formation, −1,779.6 ± 1.9 kJ/mol, was obtained by high temperature oxide melt drop solution calorimetry.
Decomposition of synthetic metastudtite was characterized by thermogravimetry and differential scanning calorimetry (DSC)
with ex situ X-ray diffraction analysis. Four decomposition steps were observed in oxygen atmosphere: water loss around 220
°C associated with an endothermic heat effect accompanied by amorphization; another water loss from 400 °C to 530 °C; oxygen
loss from amorphous UO3 to crystallize orthorhombic α-UO2.9; and reduction to crystalline U3O8. This detailed characterization allowed calculation of formation enthalpy from heat effects on decomposition measured by
DSC and by transposed temperature drop calorimetry, and both these values agree with that from drop solution calorimetry.
The data explain the irreversible transformation from studtite to metastudtite, the conditions under which metastudtite may
form, and its significant role in the oxidation, corrosion, and dissolution of nuclear fuel in contact with water.
Chemisorption of water onto anhydrous nanophase manganese oxide surfaces promotes rapidly reversible redox phase changes as
confirmed by calorimetry, X-ray diffraction, and titration for manganese average oxidation state. Surface reduction of bixbyite
(Mn2O3) to hausmannite (Mn3O4) occurs in nanoparticles under conditions where no such reactions are seen or expected on grounds of bulk thermodynamics
in coarse-grained materials. Additionally, transformation does not occur on nanosurfaces passivated by at least 2% coverage
of what is likely an amorphous manganese oxide layer. The transformation is due to thermodynamic control arising from differences
in surface energies of the two phases (Mn2O3 and Mn3O4) under wet and dry conditions. Such reversible and rapid transformation near room temperature may affect the behavior of
manganese oxides in technological applications and in geologic and environmental settings.
Co-reporter:Di Wu ; Jeremiah J. Gassensmith ; Douglas Gouvêa ; Sergey Ushakov ; J. Fraser Stoddart
Journal of the American Chemical Society 2013 Volume 135(Issue 18) pp:6790-6793
Publication Date(Web):April 23, 2013
DOI:10.1021/ja402315d
The enthalpy of adsorption of CO2 on an environmentally friendly metal–organic framework, CD-MOF-2, has been determined directly for the first time using adsorption calorimetry at 25 °C. This calorimetric methodology provides a much more accurate and model-independent measurement of adsorption enthalpy than that obtained by calculation from the adsorption isotherms, especially for systems showing complex and strongly exothermic adsorption behavior. The differential enthalpy of CO2 adsorption shows enthalpy values in line with chemisorption behavior. At near-zero coverage, an irreversible binding event with an enthalpy of −113.5 kJ/mol CO2 is observed, which is followed by a reversible −65.4 kJ/mol binding event. These enthalpies are assigned to adsorption on more and less reactive hydroxyl groups, respectively. Further, a second plateau shows an enthalpy of −40.1 kJ/mol and is indicative of physisorbed CO2. The calorimetric data confirm the presence of at least two energetically distinct binding sites for chemisorbed CO2 on CD-MOF-2.
Co-reporter:Lili Wu, Alexandra Navrotsky, Yongmoon Lee, Yongjae Lee
Microporous and Mesoporous Materials 2013 Volume 167() pp:221-227
Publication Date(Web):February 2013
DOI:10.1016/j.micromeso.2012.09.003
A series of synthetic natrolites with different non-framework cations (Li-NAT, Na-NAT, K-NAT, Rb-NAT, Cs-NAT, Ca-NAT, Sr-NAT and Pb-NAT) are investigated by high temperature oxide melt solution calorimetry. The formation enthalpy from the component oxides (ΔHf,ox) becomes less exothermic with increasing ionic potential (Z/r) of the cations. The dehydration behavior of NATs is examined using TG–DSC. The strength of water binding decreases with increasing cation size. Similar to the trend seen in anhydrous zeolites, a linear dependence of ΔHf,ox on Al/(Al + Si) ratio for hydrous zeolites is established.Graphical abstractHighlights► Enthalpies of formation of alkali and alkaline earth cation exchanged natrolites were measured. ► The results show systematic trends with cation type and Al/(Al + Si). ► The energetics of hydration and tendency toward amorphization were studied. ► A model for enthalpies of formation for hydrous aluminosilicate zeoiltes has been established.
Co-reporter:S. Mahboobeh Hosseini, Tatiana Shvareva, Alexandra Navrotsky
Solid State Ionics 2013 Volume 233() pp:62-66
Publication Date(Web):21 February 2013
DOI:10.1016/j.ssi.2012.12.012
Lanthanum silicate apatite materials, which are good ionic conductors, were prepared and characterized in two series, LSO: La9.33 + x(SiO4)6O2 + 3x / 2 (x = 0, 0.33 and 0.67) and LSSO: La10 − xSrx(SiO4)6O3 − 0.5x (x = 1.00 and 2.00). Enthalpies of formation from their binary oxides at 298 K were determined by high temperature oxide melt solution calorimetry. The energetics of these materials is discussed in terms of the effects of cation vacancies and interstitial oxygens. The formation of LSO apatites becomes more exothermic as the number of cation vacancies decreases (oxygen excess increases). Cation vacancy content is the dominant factor in determining the energetics of La9.33 + x(SiO4)6O2 + 3x / 2. The endothermic enthalpy of defect formation is 133.8 ± 17.5 kJ per mole of interstitial oxide ions, and 272.2 ± 21.6 kJ per mole of cation vacancies. Thus, La8Sr2(SiO4)6O2, having neither vacancies nor interstitial oxide ions, is the most stable compound in these two series.Highlights► Energetics of La9.33+x(SiO4)6O2+3x/2 and La10-xSrx(SiO4)6O3-0.5x are investigated. ► Stoichiometric sample La8Sr2(SiO4)6O2 is the most stable composition. ► ∆H°f, interstitial and ΔHf, cationvacancy were determined. ► Cation vacancy concentrations appear to be the dominant factor in energetics. ► Energetics in LSSO series directly correlates with conductivity.
Co-reporter:Sabyasachi Sen;Scarlett J. Widgeon;Gabriela Mera;Amir Tavakoli;Emanuel Ionescu;Ralf Riedel;
Proceedings of the National Academy of Sciences 2013 110(40) pp:15904-15907
Publication Date(Web):September 16, 2013
DOI:10.1073/pnas.1312771110
Amorphous silicon oxycarbide polymer-derived ceramics (PDCs), synthesized from organometallic precursors, contain carbon-
and silica-rich nanodomains, the latter with extensive substitution of carbon for oxygen, linking Si-centered SiOxC4-x tetrahedra. Calorimetric studies demonstrated these PDCs to be thermodynamically more stable than a mixture of SiO2, C, and silicon carbide. Here, we show by multinuclear NMR spectroscopy that substitution of C for O is also attained in
PDCs with depolymerized silica-rich domains containing lithium, associated with SiOxC4-x tetrahedra with nonbridging oxygen. We suggest that significant (several percent) substitution of C for O could occur in
more complex geological silicate melts/glasses in contact with graphite at moderate pressure and high temperature and may
be thermodynamically far more accessible than C for Si substitution. Carbon incorporation will change the local structure
and may affect physical properties, such as viscosity. Analogous carbon substitution at grain boundaries, at defect sites,
or as equilibrium states in nominally acarbonaceous crystalline silicates, even if present at levels at 10–100 ppm, might
form an extensive and hitherto hidden reservoir of carbon in the lower crust and mantle.
Co-reporter:Amir H. Tavakoli, Pardha Saradhi Maram, Scarlett J. Widgeon, Jorgen Rufner, Klaus van Benthem, Sergey Ushakov, Sabyasachi Sen, and Alexandra Navrotsky
The Journal of Physical Chemistry C 2013 Volume 117(Issue 33) pp:17123-17130
Publication Date(Web):July 22, 2013
DOI:10.1021/jp405820g
To provide a complete picture of the energy landscape of Al2O3 at the nanoscale, we directed this study toward understanding the energetics of amorphous alumina (a-Al2O3). a-Al2O3 nanoparticles were obtained by condensation from gas phase generated through laser evaporation of α-Al2O3 targets in pure oxygen at25 Pa. As-deposited nanopowders were heat-treated at different temperatures up to 600 °C to provide powders with surface areas of 670–340 m2/g. The structure of the samples was characterized by powder X-ray diffraction, transmission electron microscopy, and solid-state nuclear magnetic resonance spectroscopy. The results indicate that the microstructure consists of aggregated 3–5 nm nanoparticles that remain amorphous to temperatures as high as 600 °C. The structure consists of a network of AlO4, AlO5, and AlO6 polyhedra, with AlO5 being the most abundant species. The presence of water molecules on the surfaces was confirmed by mass spectrometry of the gases evolved on heating the samples under vacuum. A combination of BET surface-area measurements, water adsorption calorimetry, and high-temperature oxide melt solution calorimetry was employed for thermodynamic analysis. By linear fit of the measured excess enthalpy of the nanoparticles as a function of surface area, the surface energy of a-Al2O3 was determined to be 0.97 ± 0.04 J/m2. We conclude that the lower surface energy of a-Al2O3 compared with crystalline polymorphs γ- and α-Al2O3 makes this phase the most energetically stable phase at surface areas greater than 370 m2/g.
Co-reporter:Manas K. Bhunia, James T. Hughes, James C. Fettinger, and Alexandra Navrotsky
Langmuir 2013 Volume 29(Issue 25) pp:8140-8145
Publication Date(Web):May 31, 2013
DOI:10.1021/la4012839
Metal–organic framework (MOF) porosity relies upon robust metal–organic bonds to retain structural rigidity upon solvent removal. Both the as-synthesized and activated Cu and Zn polymorphs of HKUST-1 were studied by room temperature acid solution calorimetry. Their enthalpies of formation from dense assemblages (metal oxide (ZnO or CuO), trimesic acid (TMA), and N,N-dimethylformamide (DMF)) were calculated from the calorimetric data. The enthalpy of formation (ΔHf) of the as-synthesized Cu-HKUST-H2O ([Cu3TMA2·3H2O]·5DMF) is −52.70 ± 0.34 kJ per mole of Cu. The ΔHf for Zn-HKUST-DMF ([Zn3TMA2·3DMF]·2DMF) is −54.22 ± 0.57 kJ per mole of Zn. The desolvated Cu-HKUST-dg [Cu3TMA2] has a ΔHf of 16.66 ± 0.51 kJ/mol per mole Cu. The ΔHf for Zn-HKUST-amorph [Zn3TMA2·2DMF] is −3.57 ± 0.21 kJ per mole of Zn. Solvent stabilizes the Cu-HKUST-H2O by −69.4 kJ per mole of Cu and Zn-HKUST-DMF by at least −50.7 kJ per mole of Zn. Such strong chemisorption of solvent is similar in magnitude to the strongly exothermic binding at low coverage for chemisorbed H2O on transition metal oxide nanoparticle surfaces. The strongly exothermic solvent–framework interaction suggests that solvent can play a critical role in obtaining a specific secondary building unit (SBU) topology.
Co-reporter:Nancy Birkner;William H. Casey;Sara Nayeri;Mohammad Mahdi Najafpour;Babak Pashaei
PNAS 2013 Volume 110 (Issue 22 ) pp:8801-8806
Publication Date(Web):2013-05-28
DOI:10.1073/pnas.1306623110
Previous measurements show that calcium manganese oxide nanoparticles are better water oxidation catalysts than binary manganese
oxides (Mn3O4, Mn2O3, and MnO2). The probable reasons for such enhancement involve a combination of factors: The calcium manganese oxide materials have
a layered structure with considerable thermodynamic stability and a high surface area, their low surface energy suggests relatively
loose binding of H2O on the internal and external surfaces, and they possess mixed-valent manganese with internal oxidation enthalpy independent
of the Mn3+/Mn4+ ratio and much smaller in magnitude than the Mn2O3-MnO2 couple. These factors enhance catalytic ability by providing easy access for solutes and water to active sites and facile
electron transfer between manganese in different oxidation states.
Co-reporter:James T. Hughes ; Thomas D. Bennett ; Anthony K. Cheetham
Journal of the American Chemical Society 2012 Volume 135(Issue 2) pp:598-601
Publication Date(Web):December 27, 2012
DOI:10.1021/ja311237m
The first thermochemical analysis by room-temperature aqueous solution calorimetry of a series of zeolite imidazolate frameworks (ZIFs) has been completed. The enthalpies of formation of the evacuated ZIFs—ZIF-zni, ZIF-1, ZIF-4, CoZIF-4, ZIF-7, and ZIF-8—along with as-synthesized ZIF-4 (ZIF-4·DMF) and ball-milling amorphized ZIF-4 (amZIF-4) were measured with respect to dense components: metal oxide (ZnO or CoO), the corresponding imidazole linker, and N,N dimethylformamide (DMF) in the case of ZIF-4·DMF. Enthalpies of formation of ZIFs from these components at 298 K are exothermic, but the ZIFs are metastable energetically with respect to hypothetical dense components in which zinc is bonded to nitrogen rather than oxygen. These enthalpic destabilizations increase with increasing porosity and span a narrow range from 13.0 to 27.1 kJ/mol, while the molar volumes extend from 135.9 to 248.8 cm3/mol; thus, almost doubling the molar volume results in only a modest energetic destabilization. The experimental results are supported by DFT calculations. The series of ZIFs studied tie in with previously studied MOF-5, creating a broader trend that mirrors a similar pattern by porous inorganic oxides, zeolites, zeotypes, and mesoporous silicas. These findings suggest that no immediate thermodynamic barrier precludes the further development of highly porous materials.
Chemistry of Materials 2012 Volume 24(Issue 12) pp:2311
Publication Date(Web):June 7, 2012
DOI:10.1021/cm3005198
Enthalpies of formation of Cox Zn1–x O solid solutions (both bulk and nanophase materials) at 298 K have been determined using high-temperature oxide melt solution calorimetry in molten sodium molybdate (3Na2O·4MoO3) solvent at 973 K. Both the rocksalt and wurtzite phases show an approximately linear dependence of enthalpy of solution on composition, implying a zero heat of mixing in each phase, consistent with negligible lattice parameter changes on substitution of Co2+ for Zn2+. The surface energy of wurtzite Zn0.88Co0.12O solid solution was determined to be 2.33 ± 0.30 J/m2 (anhydrous surface) and 1.65 ± 0.25 J/m2 (hydrous surface), which are very close to values for ZnO. The wurtzite CoO surface energy was estimated to be similar. Here, we argue that, because of the lower surface energies of wurtzite phases than of rocksalt phases, the phase field of the wurtzite solid solution expands to higher CoO content at the nanoscale, suggesting that the reported extended solubility of CoO in ZnO nanoparticles represents thermodynamic stabilization and free energy minimization at the nanoscale. Conversely, the rocksalt Co1–xZnxO phase shows thermodynamic destabilization, lower zinc content, and easier oxidation (to Co3–xZnxO4 spinel phase) at the nanoscale than in the bulk.Keywords: CoO; nanoparticles; solid solution; surface energy; thermodynamics; ZnO;
Chemistry of Materials 2012 Volume 24(Issue 21) pp:4185
Publication Date(Web):October 2, 2012
DOI:10.1021/cm302446e
With ionic conductivities superior to conventional doped zirconia and ceria at intermediate temperatures (IT, 700–800 °C), bismuth oxide (BiO1.5) materials based on the defect fluorite structure are promising electrolyte candidates for solid oxide fuel cells (SOFCs) operating at reduced temperatures. In order to investigate the energetics of stabilized BiO1.5 in the fluorite structure, DyO1.5-stabilized BiO1.5 (DSB) over a range of compositions was synthesized by solid state reaction and quenched. Using high temperature oxide melt solution calorimetry in molten 3Na2O·4MoO3 at 702 °C, enthalpies of formation at 25 °C were determined. Relative to the phases of the oxide end-members stable at room temperature (monoclinic BiO1.5 and C-type DyO1.5), the formation of Bi1-xDyxO1.5 is endothermic at x < 0.30 and becomes slightly exothermic toward the upper phase boundary (x = 0.50). These data suggest that this system is slightly stabilized, and there is only a moderate (1–2 orders of magnitude) decrease in the volatility and susceptibility to reduction of bismuth oxide at high temperatures. However, high conductivity still makes the system potentially useful at 700 °C and below. Similar to findings for rare earth-doped zirconia, hafnia, and ceria, a negative interaction parameter for mixing in the solid solution suggests a tendency for short-range ordering, and the increasingly exothermic ΔHmix with increasing x parallels the conductivity decrease with increasing dopant content.Keywords: bismuth oxide; fluorite; solid oxide fuel cells; thermodynamics;
Co-reporter:A. V. Radha, J. D. Furman, M. Ati, B. C. Melot, J. M. Tarascon and A. Navrotsky
Journal of Materials Chemistry A 2012 vol. 22(Issue 46) pp:24446-24452
Publication Date(Web):29 Aug 2012
DOI:10.1039/C2JM34071B
Isothermal acid solution calorimetry was employed to investigate the relative thermochemical stabilities of two polymorphs of the LiFe1−xMnxSO4F (0 ≤ x ≤ 1) solid solution series: triplite and tavorite. These compounds have shown promise as lithium-ion battery cathodes, and a fuller understanding of their thermodynamics will aid in synthesis and their practical application. The linear energetic trends among triplites and tavorites indicate greater stabilization of each of these structures from the binary components with increase in manganese content and suggest a negligible heat of mixing of Fe and Mn ions. The tavorite phase, formed for x < 0.2, appears energetically more stable than the triplite. The formation of the disordered triplite structure appears to be entropy driven, and the factors that increase the disorder of the system (e.g. rapid phase formation) favor the triplite structure. Further, the free energy change associated with the tavorite to triplite transformation obtained by calculating configurational entropies using measured enthalpies was almost zero (−1.3 ± 0.8 kJ mol−1) at ambient temperature but becomes exothermic at 500 °C (−4.3 ± 0.8 to −6.8 ± 0.8 kJ mol−1). This suggests that both tavorite and triplite (with random cation distribution) are equally stable at ambient temperature but the tavorite to triplite transformation is thermodynamically favored at high temperatures because of entropy.
The reason for the higher thermal persistence of amorphous polymer-derived SiBCN ceramics (T ∼ 1700–2000 °C) compared to SiCN ones (T ∼ 1500 °C) has been a matter of debate for more than a decade. Despite recent experimental results which indicate a major kinetic effect of boron on the thermal persistence of the ceramics, no experimental investigation of the thermodynamic stability of the materials has been reported. In this work, we present measured energetics of a series of the amorphous ceramics with various boron contents (0–8.3 at.%) using high-temperature oxidative drop-solution calorimetry. Through measurement of the drop-solution enthalpies in molten sodium molybdate at 811 °C, the formation enthalpies of the amorphous ceramics from crystalline components (SiC, BN, Si3N4, C) at 25 °C were obtained and found to be between −1.4 and −26.6 kJ g-atom−1. The determined enthalpy data plus the estimated positive entropy of formation values point to the thermodynamic stability of the amorphous ceramics relative to the crystalline phases, but such stabilization diminishes with increasing boron content. In contrast, the higher boron content increases the temperature of Si3N4 crystallization despite less favorable energetics for the amorphous phase, implying more favorable energetics for crystallization. Thus the so-called “stability” of SiBCN ceramics in terms of persistence against Si3N4 crystallization appears to be controlled by kinetics rather than by thermodynamic stability.
The capacity to incorporate actinide cations makes pyrochlore titanates first-choice phases in titanate-based waste form ceramics. Despite broad interest in the pyrochlore order–disorder transformation due to the cumulative effects of 238U, 235U and 232Th radioactive decay and their daughter products, only limited thermodynamic data, mainly based on simulations of ion-beam irradiation experiments, have been reported. In this work, for the first time, heavily disordered pyrochlores, RE2Ti2O7 (RE = Y, Gd and Dy), from mechanical milling of their constituent oxides, were thermochemically investigated. Two types of thermal events were identified using high-temperature differential scanning calorimetry and correlated to the structural disorder in the cation and anion sublattices. Moreover, the excess formation energy measured by oxide melt solution calorimetry shows that the smaller the ionic radius of the RE, the easier it is to remove damage domains.
Co-reporter:Benjamin E. Hanken, Tatiana Y. Shvareva, Niels Grønbech-Jensen, Christopher R. Stanek, Mark Asta and Alexandra Navrotsky
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 16) pp:5680-5685
Publication Date(Web):05 Mar 2012
DOI:10.1039/C2CP40295E
Cation mixing energetics in urania–ceria solid solutions with stoichiometric oxygen concentrations (U1−yCeyO2) have been measured by high-temperature oxide-melt drop-solution calorimetry. Measurements have been performed on eight samples with compositions spanning y = 0.119 to y = 0.815. The measured mixing enthalpies (ΔHmix) range from −0.6 ± 3.3 to 3.9 ± 3.0 kJ mol−1. These values are discussed in the context of results from atomistic modeling which take into consideration the possibility of charge transfer between uranium and cerium cations to form solid solutions with mixed charge states. A comparison between measured and calculated results for ΔHmix suggests that such charge transfer takes place to a limited extent in the most concentrated mixtures studied.
Co-reporter:M. P. Saradhi, S. V. Ushakov and A. Navrotsky
RSC Advances 2012 vol. 2(Issue 8) pp:3328-3334
Publication Date(Web):24 Feb 2012
DOI:10.1039/C2RA00727D
The energetics of the order-disorder phase transformation in the binary oxide system, Eu2O3–ZrO2, is studied by powder X-ray diffraction and high temperature drop solution calorimetry. The nanocrystalline defect fluorite phase of Eu2Zr2O7 is synthesized on crystallization of an amorphous precursor from aqueous precipitation. The defect fluorite transforms to an ordered pyrochlore above 1200 °C. Aerodynamic levitation combined with laser heating is used to prepare coarse defect fluorite, which is otherwise impossible by conventional synthesis techniques. Formation enthalpies from oxides are −62.4 ± 2.6 and −24.6 ± 3.7 kJ mol−1 for the pyrochlore and defect fluorite phase, respectively. The transformation enthalpy from pyrochlore to defect flourite in the coarse sample is 37.8 ± 3.1 kJ mol−1 at 25 °C. The enthalpy of water vapor adsorption on the surface of the nanocrystalline defect fluorite Eu2Zr2O7 is −75 ± 2.5 kJ mol−1 H2O for coverage of 9.5 ± 0.8 H2O/nm2. The calculated surface enthalpies for the anhydrous and hydrous surfaces of defect fluorite Eu2Zr2O7 are 1.47 ± 0.13 and 1.01 ± 0.15 J m−2, respectively.
Co-reporter:Tatiana Y. Shvareva, Jeremy B. Fein, and Alexandra Navrotsky
Industrial & Engineering Chemistry Research 2012 Volume 51(Issue 2) pp:607-613
Publication Date(Web):November 12, 2011
DOI:10.1021/ie2002582
More than 50 uranyl minerals, phases containing U6+ as the uranyl UO22+ cation, and hydroxide, carbonate, phosphate, and silicate anions, H2O, and alkali and alkaline earth cations, occur in nature and as corrosion products of spent nuclear fuel. Despite their importance and the need to understand their thermodynamics to predict uranium solubility, fate, and transport in the environment, reliable thermodynamic data have only been available recently. This paper summarizes recent studies of enthalpies of formation using high temperature oxide melt solution calorimetry and Gibbs free energies from solubility experiments. Standard state thermochemical parameters (at 25 °C and 1 bar) are tabulated and the stability and transformation sequences of these phases are discussed. The enthalpies of formation from oxides are discussed in terms of crystal structure and Lewis acid–base interactions.
Co-reporter:Olga Trofymluk, Andrey A. Levchenko, Alexandra Navrotsky
Microporous and Mesoporous Materials 2012 Volume 149(Issue 1) pp:119-125
Publication Date(Web):1 February 2012
DOI:10.1016/j.micromeso.2011.08.022
The thermodynamics of mesoporous silicas (MCM-41, MCM-48, SBA-15, and SBA-16) were studied by solution calorimetry at 323 K in 25% aqueous HF. The enthalpies of formation were determined for calcined mesoporous silica (MS) and organic structure-directing agent (SDA) occluded samples (SDA: n-hexadeciltrimethylammonium bromide or CTAB, Pluronic P123, and Pluronic F127). The following are the measured interaction enthalpies between the MS and SDA: MCM-41/CTAB, −6.1 kJ/mol SiO2; MCM-48/CTAB, −12.3 kJ/mol SiO2; SBA-15/P123, −19.7 kJ/mol SiO2; SBA-16/F127, −19.9 kJ/mol SiO2. Per unit surface area, these interactions are −0.08, −0.15, −0.43, and −0.40 J/m2, respectively. Though these SDA–framework interaction energies are still small in magnitude, they are somewhat more exothermic than those in silica zeolite formation, reflecting the greater metastability of the MS materials and the role of the long chain SDA in stabilizing and space-filling the large pores. The cubic MS (SBA) show stronger SDA interactions than the hexagonal (MCM). The interaction energies confirm a complex landscape of many competing structures of similar energy; with the role of SDA kinetic in selecting a specific structure rather than energetic in strongly stabilizing a given state, as has already been noted for zeolites. The enthalpies of the calcined MS relative to quartz determined by HF solution calorimetry in this study are in excellent agreement with those determined previously by high temperature oxide melt solution calorimetry.Graphical abstractHighlights► The energetics of synthesis of MCM-41/48, SBA-15/16 have been studied. ► Enthalpies of solution in HF are measured for the as-made and calcined silicas. ► The interaction enthalpies of CTAB, Pluronic P123/F127 and hosts are calculated. ► The enthalpies of interaction span an exothermic range of −6.0 to −19.6 kJ/mol SiO2. ► Per unit surface area these interactions are negative 0.08–0.43 J/m2.
Co-reporter:Salih Buyukkilic, Tatiana Shvareva, Alexandra Navrotsky
Solid State Ionics 2012 Volume 227() pp:17-22
Publication Date(Web):29 October 2012
DOI:10.1016/j.ssi.2012.08.017
It has been suggested that co-doping ceria with two trivalent ions of different sizes to minimize lattice strain produces materials with better ionic conductivity. To investigate the thermodynamic basis of such behavior, enthalpies of formation at room temperature of samarium-doped ceria (Ce1 − xSmxO2 − 0.5x with 0 < x < 0.3), neodymium-doped ceria (Ce1 − xNdxO2 − 0.5x with 0 < x < 0.3), and neodymia–samaria co-doped ceria (Ce1 − xNdx/2Smx/2O2 − 0.5x with 0 < x < 0.3) have been measured by high temperature oxide melt solution calorimetry. The energetics of the solid solutions were analyzed in terms of cation size mismatch and defect association. At concentrations below x = 0.05, endothermic (destabilization) heat of formation is attributed to the dominance of size mismatch. Considerable energetic stabilization at x > 0.05 for singly doped ceria systems can be attributed to defect associates of trivalent cations coupled with charge-balancing oxygen vacancies. For co-doped Ce1 − xNdx/2Smx/2O2 − 0.5x, there is less destabilization at low x compared to singly doped CeO2 but less stabilization at high x and a shift in the composition of maximum (most endothermic) formation enthalpy toward higher dopant concentration. Enthalpies of defect association of Ce1 − xNdx/2Smx/2O2 − 0.5x are less exothermic than those of singly doped materials.Highlights► ΔHf,ox, ΔHmix, and ΔHassoc of xSDC, xNDC, and xSNDC are reported. ► Maximum ΔHf,ox are found 9.07 ± 1.17 kJ/mol for 5SDC and 8.95 ± 1.30 kJ/mol for 5NDC. ► Maximum ΔHf,ox, 6.13 ± 1.06 kJ/mol, is shifted toward higher x in 10SNDC. ► ΔHassoc of SNDC are less exothermic than those of SDC and NDC. ► Our findings confirm the conductivity measurements at low temperature.
Co-reporter:Christopher R. Armstrong;Ginger E. Sigmon;May Nyman;Peter C. Burns;Tatiana Shvareva
PNAS 2012 Volume 109 (Issue 6 ) pp:
Publication Date(Web):2012-02-07
DOI:10.1073/pnas.1119758109
The Fukushima-Daiichi nuclear accident brought together compromised irradiated fuel and large amounts of seawater in a high
radiation field. Based on newly acquired thermochemical data for a series of uranyl peroxide compounds containing charge-balancing
alkali cations, here we show that nanoscale cage clusters containing as many as 60 uranyl ions, bonded through peroxide and
hydroxide bridges, are likely to form in solution or as precipitates under such conditions. These species will enhance the
corrosion of the damaged fuel and, being thermodynamically stable and kinetically persistent in the absence of peroxide, they
can potentially transport uranium over long distances.
Journal of the American Chemical Society 2011 Volume 133(Issue 24) pp:9184-9187
Publication Date(Web):May 20, 2011
DOI:10.1021/ja202132h
The first experimental thermodynamic analysis of a metal–organic framework (MOF) has been performed. Measurement of the enthalpy of formation of MOF-5 from the dense components zinc oxide (ZnO), 1,4-benzenedicarboxylic acid (H2BDC), and occluded N,N-diethylformamide (DEF) (if any) gave values of 78.64 ± 2.95 and 99.47 ± 3.62 kJ·[mol of Zn4O(BDC)3·xDEF]−1 for the as-made form and the desolvated structure, respectively. These as-made and desolvated enthalpies correspond to the values 19.66 ± 0.74 and 24.87 ± 0.94 kJ·(mol of Zn)−1, respectively. The energetics of desolvated MOF-5 per mole of Zn falls in line with trends relating the enthalpy of inorganic porous materials (zeolites, zeotypes, and mesoporous materials) to molar volume. MOF-5 extends a plateauing trend first suggested by thermodynamic studies of mesoporous materials. This leveling off of the destabilization energetics as the void space swells suggests that additional void volume beyond a certain point may begin to act as a parameter “external” to the structure and not destabilize it further. This could help explain the rich landscape of large-volume MOFs and their ease of desolvation.
Co-reporter:Yin-Qing Zhang, Wei Zhou, Shuangxi Liu, and Alexandra Navrotsky
Chemistry of Materials 2011 Volume 23(Issue 5) pp:1166
Publication Date(Web):January 21, 2011
DOI:10.1021/cm103444e
Titanosilicate ETS-4 materials with three different morphologies were synthesized hydrothermally by controlling the pH of the synthesis gel mixtures. Their morphology changed from thin rectangular plate monolithic crystals to double-fan-like polycrystalline aggregates as the pH decreased. The crystallization time depended on the initial pH. The effect of morphology on photocatalytic activity was investigated using the photodegradation reaction of Rhodamine B under visible light irradiation. The double-fan-like material exhibited significantly greater photodegradation activity than the other two ETS-4 samples and Degussa P25 titania. Powder X-ray diffraction, electron microprobe analysis, scanning electron microscopy, Raman spectroscopy, and diffuse reflectance ultraviolet−visible spectroscopy were performed to find possible reasons for the photocatalytic activity difference of the three samples. The enthalpies of formation from oxides for all the ETS-4 materials were investigated by high temperature oxide melt calorimetry. The results showed that the three samples with different morphologies have differences in their enthalpies of formation, which, although small, are bigger than likely to arise from surface area effects alone, and the product with most Ti−OH groups has the smallest thermodynamic stability. The calorimetric and spectroscopic data together provide evidence that the high photocatalytic activity of the double-fan-like aggregates is linked to their high Ti−OH content.Keywords: calorimetry; enthalpy of formation; ETS-4; morphology; photocatalysis;
Co-reporter:Gustavo Carneiro Cardoso da Costa, Lili Wu and Alexandra Navrotsky
Journal of Materials Chemistry A 2011 vol. 21(Issue 6) pp:1837-1845
Publication Date(Web):08 Dec 2010
DOI:10.1039/C0JM03090B
The energetics of the (1 − x)PMN–xPT solid solutions have been investigated using high temperature oxide melt solution calorimetry in 3Na2O·4MoO3solvent at 702 °C. The solid solutions show positive heats of mixing, reflecting the changes in structure from cubic to tetragonal to monoclinic and the morphotropic phase transition. The synthesis of a perovskite phase without a secondary pyrochlore phase depends on the annealing temperature and time. The enthalpies of reactions involved in synthesis and decomposition of the PMN perovskite were measured for the first time by oxide melt solution calorimetry. The decomposition of perovskite to pyrochlore plus MgO and PbO is energetically favorable, as is the formation of perovskite from columbite and lead oxide.
Co-reporter:Andrey A. Levchenko, Alexander I. Kolesnikov, Olga Trofymluk, Alexandra Navrotsky
Carbon 2011 Volume 49(Issue 3) pp:949-954
Publication Date(Web):March 2011
DOI:10.1016/j.carbon.2010.11.004
Bundles of (10,10) single-wall carbon nanotubes (SWCNTs) have been studied by high-temperature oxidation calorimetry and inelastic neutron scattering to obtain standard formation enthalpies and entropies at 298 K. SWCNTs are found to be only moderately less stable than graphite, and are significantly more stable than their fullerene counterparts. They are 7 kJ mol−1 metastable in terms of enthalpy relative to graphite, and just 5 kJ mol−1 less stable than diamond. Despite striking differences in vibrational dynamics of carbon atoms in SWCNTs and graphite, their thermodynamic properties at room and higher temperatures are dominated by the same set of high energy vibrations, reflected in very similar vibrational entropies. However, the energetics of SWCNTs are governed by the diameter-dependent enthalpic contributions, but not the specifics of phonon density of states.Graphical abstractThis study places SWCNTs (red) on the energy landscape for carbon allotropes which include graphite, diamond and fullerenes. The enthalpic contributions control the energetics of SWCNTs, making CNTs 7 kJ mol−1 less stable than graphite. The height of the 3D blocks in the diagram shows experimental errors. The lines and rectangle are theoretical values and their span.Research highlights► Enthalpic factors rather than entropy contributions dominate the energetics of SWCNTs. ► Heat capacity and entropy of SWCNTs are similar to those of graphite above 100 K. ► SWCNTs are 7 kJ mol−1 metastable in enthalpy relative to graphite (5 kJ mol−1 to diamond). ► Direct measurements of SWCNT energetics agree reasonably with theory predictions.
Co-reporter:Wei Zhou, Pingping Sun, Peng Zhang, Alexandra Navrotsky
Microporous and Mesoporous Materials 2011 Volume 142(2–3) pp:749-753
Publication Date(Web):July 2011
DOI:10.1016/j.micromeso.2011.01.019
The thermochemistry of proton containing borosilicate, aluminosilicate and gallosilicate zeolites with beta topology (B-BEA, Al-BEA and Ga-BEA) is described. Thermogravimetry and differential scanning calorimetry on these materials indicate the substitution of Al by Ga or B in the beta framework reduces the decomposition temperature. Water adsorption calorimetry directly measured the hydration enthalpies of these samples. B-BEA and Ga-BEA have less exothermic hydration enthalpies than Al-BEA. High temperature oxide melt solution calorimetry was performed to derive the formation enthalpies of hydrated samples (8.9–18.8 kJ/mol relative to oxides on TO2 molar basis). The formation enthalpies of dehydrated phases (33.2–55.1 kJ/mol relative to oxides on TO2 molar basis) were calculated from the formation enthalpies of hydrated phases and the hydration enthalpy.Graphical abstractResearch highlights► Hydration enthalpy of zeolite beta was measured by water adsorption calorimetry. ► Formation enthalpy of zeolite beta was measured by solution calorimetry. ► The substitution of Al by B and Ga in zeolite beta affects the thermal stability.
Co-reporter:Tatiana Y. Shvareva, Vitaly Alexandrov, Mark Asta, Alexandra Navrotsky
Journal of Nuclear Materials 2011 Volume 419(1–3) pp:72-75
Publication Date(Web):December 2011
DOI:10.1016/j.jnucmat.2011.08.002
Mixing enthalpies (ΔHmix) of ThO2–CeO2 solid solutions with respect to cubic fluorite ThO2 and CeO2 have been measured by high temperature oxide melt solution calorimetry. The system shows a slightly positive mixing enthalpy, with a maximum value of ΔHmix = 3.7 ± 2.5 kJ/mol at 50% Ce/(Ce + Th). Based on the regular-solution model, with an interaction parameter of 15.1 ± 2.2 kJ/mol fit to the measured data, the phase diagram is predicted to feature a miscibility gap with a calculated critical temperature of 908 ± 132 K. The results are complemented by density-functional-theory and Monte-Carlo calculations, which provide positive mixing enthalpies and a miscibility-gap phase diagram, in qualitative agreement with calorimetric results. The calculations suggest small effects of short-range order (clustering) on the mixing enthalpy above the miscibility gap. The calculated values of ΔHmix are within the error bars of the measured values, but consistently smaller in magnitude. An analysis of the calculated results indicates that the dominant contribution to the mixing enthalpy arises from the elastic energy associated with cation size mismatch, allowing predictions of the behavior in ThO2–UO2 and ThO2–PuO2 systems. The analysis also suggests that the slightly smaller values of the computed ΔHmix relative to experiment can be attributed to an underestimation of the magnitude of the elastic moduli in the calculations.Highlights► ΔHmix of ThO2–CeO2 is measured. ► Maximum mixing enthalpy is 3.7 ± 2.5 kJ/mol at Th0.5Ce0.5O2. ► DFT and Monte-Carlo calculations are in agreement with experimental results. ► Phase diagram with critical temperature 908 ± 132 K is predicted. ► Findings allow the prediction of the behavior in ThO2–UO2 and ThO2–PuO2.
Co-reporter:Vitaly Alexandrov, Tatiana Y. Shvareva, Shmuel Hayun, Mark Asta, and Alexandra Navrotsky
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 24) pp:3130-3134
Publication Date(Web):November 30, 2011
DOI:10.1021/jz201458x
A comprehensive understanding of chemical interactions between water and actinide dioxide surfaces is critical for safe operation and storage of nuclear fuels. Despite substantial previous research, understanding the nature of these interactions remains incomplete. In this work, we combine accurate calorimetric measurements with first-principles computational studies to characterize surface energies and adsorption enthalpies of water on two fluorite-structured compounds, ThO2 and CeO2, that are relevant for understanding the behavior of water on actinide oxide surfaces more generally. We determine coverage-dependent adsorption enthalpies and demonstrate a mixed molecular and dissociative structure for the first hydration layer. The results show a correlation between the magnitude of the anhydrous surface energy and the water adsorption enthalpy. Further, they suggest a structural model featuring one adsorbed water molecule per one surface cation on the most stable facet that is expected to be a common structural signature of water adsorbed on actinide dioxide compounds.Keywords: calorimetry; density functional theory; fluorite oxides; water adsorption;
Because different solid materials (phases) have different surface energies, equilibria among them will be significantly affected by particle size. This Minireview summarizes experimental (calorimetric) data for the surface energies of oxides and discusses shifts in the stability of polymorphs, the thermodynamics of hydration, and oxidation–reduction reactions in nanoscale oxide systems.
Co-reporter:Wei Zhou, Quan Shi, Brian F. Woodfield, Alexandra Navrotsky
The Journal of Chemical Thermodynamics 2011 Volume 43(Issue 6) pp:970-973
Publication Date(Web):June 2011
DOI:10.1016/j.jct.2011.02.002
The low temperature (2 to 300) K heat capacity of monoclinic hafnia (HfO2) was measured using the heat capacity option of a Quantum Design Physical Property Measurement System (PPMS). The thermodynamic functions in this temperature range were derived by curve fitting. The standard entropy and enthalpy of hafnia at T = 298.15 K was calculated to be 56.15 ± 0.57 J · mol−1 · K−1 and 9.34 ± 0.09 kJ · mol−1, respectively. The results are in fairly good agreement with old data, which only covered temperatures from (50 to 298) K. Hafnia has a higher heat capacity than zirconia at all temperatures from (2 to 300) K.Research highlights► PPMS was successfully used to measure the low temperature Cp of hafnia. ► Thermodynamic functions at temperature from (2 to 300) K were derived. ► Comparisons between the present work and literature, hafnia and zirconia, were made.
Co-reporter:Christopher R. Armstrong;William H. Casey
PNAS 2011 Volume 108 (Issue 36 ) pp:
Publication Date(Web):2011-09-06
DOI:10.1073/pnas.1111243108
The ϵ-Al13 Keggin aluminum hydroxide clusters are essential models in establishing molecular pathways for geochemical reactions. Enthalpies
of formation are reported for two salts of aluminum centered ϵ-Keggin clusters, Al13 selenate, (Na(AlO4)Al12(OH)24(SeO4)4•12H2O) and Al13 sulfate, (NaAlO4Al12(OH)24(SO4)4•12H2O). The measured enthalpies of solution, ΔHsol, at 28 °C in 5 N HCl for the ε-Al13 selenate and sulfate are −924.57 (± 3.83) and −944.30 ( ± 5.66) kJ·mol-1, respectively. The enthalpies of formation from the elements, ΔHf,el, for Al13 selenate and sulfate are −19,656.35 ( ± 67.30) kJ·mol-1, and −20,892.39 ( ± 70.01) kJ·mol-1, respectively. In addition, ΔHf,el for sodium selenate decahydrate was calculated using data from high temperature oxide melt solution calorimetry measurements:
−4,006.39 ( ± 11.91) kJ·mol-1. The formation of both ε-Al13 Keggin cluster compounds is exothermic from oxide-based components but energetically unfavorable with respect to a gibbsite-based
assemblage. To understand the relative affinity of the ϵ-Keggin clusters for selenate and sulfate, the enthalpy associated
with two S-Se exchange reactions was calculated. In the solid state, selenium is favored in the Al13 compound relative to the binary chalcogenate, while in 5 N HCl, sulfur is energetically favored in the cluster compound compared
to the aqueous solution. This contribution represents the first thermodynamic study of ε-Al13 cluster compounds and establishes a method for other such molecules, including the substituted versions that have been created
for kinetic studies. Underscoring the importance of ε-Al13 clusters in natural and anthropogenic systems, these data provide conclusive thermodynamic evidence that the Al13 Keggin cluster is a crucial intermediate species in the formation pathway from aqueous aluminum monomers to aluminum hydroxide
precipitates.
Co-reporter:Tae-Jin Park, Andrey A. Levchenko, Hongjun Zhou, Stanislaus S. Wong and Alexandra Navrotsky
Journal of Materials Chemistry A 2010 vol. 20(Issue 39) pp:8639-8645
Publication Date(Web):08 Sep 2010
DOI:10.1039/C0JM02192J
We report the direct determination of surface enthalpies for nanophase TiO2 anatase with different morphologies derived from drop solution calorimetry in a molten sodium molybdate (3Na2Oŀ4MoO3) solvent at 702 °C. The energetics of surface hydration has been measured using a Calvet microcalorimeter coupled with a gas dosing system. The surface enthalpies of hydrated surfaces for anatase TiO2 nanoparticles, nanowires and sea-urchin-like assemblies are 0.51 ± 0.05, 1.07 ± 0.28, and 1.29 ± 0.16 J m−2, respectively, whereas those of anhydrous surfaces are 0.74 ± 0.04, 1.24 ± 0.28, and 1.41 ± 0.16 J m−2, respectively. The trend in TiO2, which shows higher surface enthalpies for more complex nanostructures, is consistent with that reported in ZnO. The shape-dependent surface enthalpy at the nanoscale level is discussed in terms of exposed surface structures. The enthalpies of hydration appear to be similar for all morphologies.
Co-reporter:Wei Zhou, Alexandra Navrotsky, Jiho Shin, Suk Bong Hong
Microporous and Mesoporous Materials 2010 Volume 135(1–3) pp:197-200
Publication Date(Web):November 2010
DOI:10.1016/j.micromeso.2010.07.004
The thermochemistry of eight gallosilicate zeolites with the NAT topology, six of which are characterized by similar Ga contents (Si/Ga ∼1.6) but different T-atom distributions and the other two materials by an unusual higher Ga content (Si/Ga ∼1.3), is described. The formation enthalpies of the sodium form of gallosilicate natrolites with lower Ga contents (Na-NAT-I, Na-NAT-II and Na-NAT-III) from oxides range from −50.3 to −57.0 kJ mol−1 of TO2 (T = Si or Ga), while those of the potassium form (K-NAT-I, K-NAT-II and K-NAT-III) lie between −65.5 and −68.4 kJ mol−1 of TO2. These small energy differences provide a thermodynamic explanation for the in situ transformation between disordered and ordered structures in the crystallization medium. While the formation enthalpy of another potassium natrolite with a high Ga content (K-PST-1) is highly exothermic, consistent with its high thermal stability, its sodium counterpart (Na-PST-1) has a considerable less exothermic formation enthalpy, as well as lower thermal stability.
Co-reporter:Tori Z. Forbes, May Nyman, Mark A. Rodriguez, Alexandra Navrotsky
Journal of Solid State Chemistry 2010 Volume 183(Issue 11) pp:2516-2521
Publication Date(Web):November 2010
DOI:10.1016/j.jssc.2010.08.024
Lanthanum tantalates are important refractory materials with application in photocatalysis, solid oxide fuel cells, and phosphors. Soft-chemical synthesis utilizing the Lindqvist ion, [Ta6O19]8−, has yielded a new phase, La2Ta2O7(OH)2. Using the hydrated phase as a starting material, a new lanthanum orthotantalate polymorph was formed by heating to 850 °C, which converts to a previously reported LaTaO4 polymorph at 1200 °C. The stabilities of La2Ta2O7(OH)2 (LaTa−OH), the intermediate LaTaO4 polymorph (LaTa-850), and the high temperature phase (LaTa-1200) were investigated using high-temperature oxide melt solution calorimetry. The enthalpy of formation from the oxides were calculated from the enthalpies of drop solution to be −87.1±9.6, −94.9±8.8, and −93.1±8.7 kJ/mol for LaTa−OH, LaTa-850, and LaTa-1200, respectively. These results indicate that the intermediate phase, LaTa-850, is the most stable. This pattern of energetics may be related to cation–cation repulsion of the tantalate cations. We also investigated possible LnTaO4 and Ln2Ta2O7(OH)2 analogues of Ln=Pr, Nd to examine the relationship between cation size and the resulting phases.Graphical abstractThe energetics of three lanthanum tantalates were investigated by the high-temperature oxide melt solution calorimetry. The enthalpies of formation from the oxides were calculated from the enthalpies of drop solution to be −87.1±9.6, −94.9±8.8, and −93.1±8.7 kJ/mol for La2Ta2O7(OH)2, LaTaO4 (850 °C), and LaTaO4 (1200 °C), respectively. These results indicate that the intermediate phase, LaTaO4 (850 °C), is the most stable in energy.
Co-reporter:Christopher E. Killian;A. V. Radha;Tori Z. Forbes;P. U. P. A. Gilbert
PNAS 2010 Volume 107 (Issue 38 ) pp:16438-16443
Publication Date(Web):2010-09-21
DOI:10.1073/pnas.1009959107
Amorphous calcium carbonate (ACC) is a metastable phase often observed during low temperature inorganic synthesis and biomineralization.
ACC transforms with aging or heating into a less hydrated form, and with time crystallizes to calcite or aragonite. The energetics
of transformation and crystallization of synthetic and biogenic (extracted from California purple sea urchin larval spicules,
Strongylocentrotus purpuratus) ACC were studied using isothermal acid solution calorimetry and differential scanning calorimetry. Transformation and crystallization
of ACC can follow an energetically downhill sequence: more metastable hydrated ACC → less metastable hydrated ACC⇒anhydrous
ACC ∼ biogenic anhydrous ACC⇒vaterite → aragonite → calcite. In a given reaction sequence, not all these phases need to occur.
The transformations involve a series of ordering, dehydration, and crystallization processes, each lowering the enthalpy (and
free energy) of the system, with crystallization of the dehydrated amorphous material lowering the enthalpy the most. ACC
is much more metastable with respect to calcite than the crystalline polymorphs vaterite or aragonite. The anhydrous ACC is
less metastable than the hydrated, implying that the structural reorganization during dehydration is exothermic and irreversible.
Dehydrated synthetic and anhydrous biogenic ACC are similar in enthalpy. The transformation sequence observed in biomineralization
could be mainly energetically driven; the first phase deposited is hydrated ACC, which then converts to anhydrous ACC, and
finally crystallizes to calcite. The initial formation of ACC may be a first step in the precipitation of calcite under a
wide variety of conditions, including geological CO2 sequestration.
Co-reporter:Leah N. Appelhans ; Monica Kosa ; A. V. Radha ; Petra Simoncic ; Alexandra Navrotsky ; Michele Parrinello ;Anthony K. Cheetham
Journal of the American Chemical Society 2009 Volume 131(Issue 42) pp:15375-15386
Publication Date(Web):October 1, 2009
DOI:10.1021/ja905690t
The hydrothermal reactions of calcium, strontium, and barium with l-, meso-, and d,l-tartaric acid were examined from room temperature to 220 °C. We report the synthesis of 13 new phases and crystal structures of 11 alkaline earth tartrates, including an unusual I3O0 framework, [Ba(d,l-Tar)] (Tar = C4H4O62−), with 3-D inorganic connectivity. Each alkaline earth exhibits different phase behavior in the reactions with the three forms of tartaric acid. Calcium forms unique l-, meso-, and d,l-tartrate phases which persist to 220 °C. Strontium forms three unique phases at lower temperatures, but above 180 °C reactions with l- and d,l-tartaric acid yield the meso phase. Likewise, Ba forms three unique low-temperature phases, but above 200 °C reactions with l- and meso-tartaric acid yield the d,l phase. Computational and calorimetric studies of the anhydrous calcium phases, [Ca(l-Tar)] and [Ca(meso-Tar)], strontium phases, [Sr(l-Tar)] and [Sr(meso-Tar)], and barium phases, [Ba(l-Tar)] and [Ba(d,l-Tar)], were performed to determine relative phase stabilities and elucidate the role of thermodynamic and kinetic factors in controlling phase behavior. The computational and calorimetric results were in excellent agreement. The [Ca(meso-Tar)] phase was found to be 9.1 kJ/mol more stable than the [Ca(l-Tar)] phase by computation (total electronic energies) and 2.9 ± 1.6 kJ/mol more stable by calorimetry (enthalpies of solution). The [Sr(meso-Tar)] phase was found to be 13.4 and 8.1 ± 1.4 kJ/mol more stable than [Sr(l-Tar)] by computation and calorimetry, respectively. Finally, the [Ba(l-Tar)] phase was found to be 6.4 and 7.0 ± 1.0 kJ/mol more stable than the [Ba(d,l-Tar)] phase. Our results suggest that the calcium and strontium meso phases are the most thermodynamically stable phases in their systems over the temperature range studied. The phase transitions are controlled by relative thermodynamic stabilities but also by a kinetic factor, likely the barrier to isomerization/racemization of the tartaric acid, which is hypothesized to preclude phase transformations at lower temperatures. In the barium system we find the [Ba(l-Tar)] phase to be the most thermodynamically stable phase at low temperatures, while the [Ba(d,l-Tar)] phase becomes the thermodynamic product at high temperatures, due to a larger entropic contribution.
Co-reporter:Wei Zhou, Pingping Sun, Alexandra Navrotsky, Seok Han Kim, Suk Bong Hong
Microporous and Mesoporous Materials 2009 Volume 121(1–3) pp:200-207
Publication Date(Web):1 May 2009
DOI:10.1016/j.micromeso.2009.02.001
A series of gallosilicate materials with different framework structures (i.e., Ga–MAZ, Ga–OFF, Ga–MOR, TNU-6, and TNU-7) and different Ga contents is investigated by high temperature solution calorimetry in molten lead borate at 700 °C. The dehydration enthalpies for the fully hydrated samples transforming to the partially dehydrated samples, which have 10–20% of the original amount of water remaining, lie in the range 2.0–14.6 kJ per mole of TO2 (T = Si or Ga) and 4.3–24.1 kJ per mole of H2O. The formation enthalpies from oxides of fully hydrated samples range from −87.9 to −4.8 kJ per mole of TO2, while the formation enthalpies of anhydrous samples are estimated to range from −87.9 to +9.8 kJ per mol TO2. The dependence of enthalpies on the framework density and Ga/(Ga + Si) ratio is examined. Similar to trends in aluminosilicate zeolites, high framework density, high T3+ content and large charge balancing cations stabilize gallosilicate materials. Enthalpies of vitrification of gallosilicate crystals are estimated by integrating differential scanning calorimetry (DSC) peaks, and formation enthalpies of gallosilicate glasses are calculated.
Journal of Thermal Analysis and Calorimetry 2009 Volume 96( Issue 2) pp:353-361
Publication Date(Web):2009 May
DOI:10.1007/s10973-008-8783-y
Differential scanning and high temperature reaction calorimetry have been used to characterize a series of natural iron ore and flux samples commonly used during iron ore sintering. Most iron ore samples were shown to contain measurable quantities of goethite, with a characteristic dehydration peak in DSC and TG between 200 and 400°C. At higher temperatures, all samples decomposed to produce magnetite with an accompanying mass loss in the TG profile due to the evolution of oxygen.High temperature reaction calorimetry has been used to measure the heat of solution of the ore in the melt formed during iron ore sintering. The dehydration and calcination of iron ore and flux samples was also examined using high-temperature reaction calorimetry. The results support the DSC/TG findings.
Co-reporter:Oscar Bomatí-Miguel, Lena Mazeina, Alexandra Navrotsky and Sabino Veintemillas-Verdaguer
Chemistry of Materials 2008 Volume 20(Issue 2) pp:591
Publication Date(Web):January 1, 2008
DOI:10.1021/cm071178o
The thermodynamic properties of maghemite (γ-Fe2O3) nanoparticles with size smaller than 10 nm had not been studied previously because of particle size limitations for samples synthesized by wet chemical methods. Laser-induced pyrolysis is a well-established method of producing maghemite with particle sizes smaller than 10 nm. Maghemite nanoparticles, obtained by this method and having a size range of 2–40 nm, were fully characterized and studied by solution calorimetry. The enthalpy of water adsorption was also measured. The surface enthalpy obtained from calorimetric data for the hydrated maghemite surface is 0.57 ± 0.10 J/m2 and is in good agreement with previously reported values. The surface enthalpy for the dry, water-free surface is 0.71 ± 0.13 J/m2 and is reported for the first time. The difference in the surface enthalpy for the dry surface between α- and γ-polymorphs of Fe2O3 is similar to that between α- and γ-Al2O3. This large difference in surface enthalpy (∼1.2 J/m2) creates an energy crossover so that fine-grained hematite is metastable relative to fine-grained maghemite at particle size <15 nm.
Microporous and Mesoporous Materials 2008 Volume 111(1–3) pp:507-516
Publication Date(Web):15 April 2008
DOI:10.1016/j.micromeso.2007.08.043
Gallosilicate zeolites Ga-NaSOD, Ga-NaFAU, Ga-NaNAT, Ga-KNAT* (a mixture of Ga-KNAT and Ga-KTNU-2), Ga-KLTL and Ga-KTNU-1 have been synthesized and characterized by X-ray diffraction (XRD), microprobe analysis, thermogravimetric analysis and differential scanning calorimetry (TG-DSC). Generally, the lattice parameters increase after Ga substitution for Al, except for Ga-KNAT*. The formation and dehydration enthalpies of these zeolites were measured by high temperature oxide melt solution calorimetry. Compared to the analogous aluminosilicate zeolite of similar Si/T 3+, the gallosilicate zeolite has similar dehydration enthalpy per mole of tetrahedra, but has less endothermic dehydration enthalpy per mole of water due to the larger number of H2O molecules in the enlarged unit cell of the Ga zeolite. The dehydration enthalpy per mole of water is a monotonic function of framework density while that per mole of tetrahedra is mainly influenced by cation type. The gallosilicate zeolites have less exothermic formation enthalpies from oxide components than the analogous aluminosilicate zeolites, confirming their lower stability. The formation enthalpies of hydrated and dehydrated gallosilicate zeolites are correlated with Ga/(Ga + Si) ratio and framework density (FD).
Microporous and Mesoporous Materials 2008 Volume 109(1–3) pp:147-155
Publication Date(Web):1 March 2008
DOI:10.1016/j.micromeso.2007.04.040
Cationic variants of zeolite beta (Mg-BEA 14, Ca-BEA 14, Sr-BEA 14, Ba-BEA 14, Mg-BEA 4 and Ca-BEA 4) were achieved by alumination and ion exchange processes, and characterized by XRD, NMR and TG–DSC. Mg-BEA 14/Ca-BEA 14, Sr-BEA 14/Ba-BEA 14, and Mg-BEA 4/Ca-BEA 4 have distinct thermal behavior. The low silica zeolites Mg-BEA 4 and Ca-BEA 4 were observed to have octahedral Al atoms and less crystallinity compared with high silica Mg-BEA 14 and Ca-BEA 14. High temperature oxide melt solution calorimetry determined the dehydration enthalpy and the formation enthalpy from the constituent oxides. For alkaline earth zeolite beta with the same Si/Al ratio, both the integral dehydration enthalpy and formation enthalpy from oxides becomes more endothermic with increasing average ionic potential. Mg-BEA 4 and Ca-BEA 4 have very endothermic formation enthalpies, in both dehydrated and hydrated forms, indicating a likely thermodynamic barrier to their direct synthesis, despite the natural occurrence of their analog, tschernichite.
Co-reporter:So-Nhu Le, Alexandra Navrotsky, Valerie Pralong
Solid State Sciences 2008 Volume 10(Issue 6) pp:761-767
Publication Date(Web):June 2008
DOI:10.1016/j.solidstatesciences.2007.08.008
Differential scanning calorimetry and high temperature oxide melt solution calorimetry are used to study enthalpy of phase transition and enthalpies of formation of Cu2P2O7 and Cu3(P2O6OH)2. α-Cu2P2O7 is reversibly transformed to β-Cu2P2O7 at 338–363 K with an enthalpy of phase transition of 0.15 ± 0.03 kJ mol−1. Enthalpies of formation from oxides of α-Cu2P2O7 and Cu3(P2O6OH)2 are −279.0 ± 1.4 kJ mol−1 and −538.8 ± 2.7 kJ mol−1, and their standard enthalpies of formation (enthalpy of formation from elements) are −2096.1 ± 4.3 kJ mol−1 and −4302.7 ± 6.7 kJ mol−1, respectively. The presence of hydrogen in diphosphate groups changes the geometry of Cu(II) and decreases acid–base interaction between oxide components in Cu3(P2O6OH)2, thus decreasing its thermodynamic stability.Difference in enthalpy of formation from oxides of basic phosphates and their relative acidic compounds as a function of acidity of metal oxides.
Co-reporter:Lena Mazeina, Alexandra Navrotsky, Martha Greenblatt
Journal of Nuclear Materials 2008 Volume 373(1–3) pp:39-43
Publication Date(Web):15 February 2008
DOI:10.1016/j.jnucmat.2007.03.269
Quantitative study of thermodynamic properties of solid solutions of UO2+x with divalent and trivalent oxides is important for predicting the behavior of oxide fuel. Although early literature work measured vapor pressure in some of these solid solutions, direct calorimetric measurements of enthalpies of formation have been hampered by the refractory nature of such oxides. First measurements of the enthalpies of formation in the systems UO2+x–CaO and UO2+x–YO1.5, obtained by high-temperature oxide melt solution calorimetry, are reported. Both systems show significantly negative (exothermic) heats of formation from binary oxides (UO2, plus O2 and CaO or YO1.5, as well as from UO2 plus UO3 and CaO or YO1.5), consistent with reported free energy measurements in the urania–yttria system. The energetic contributions of oxygen content (oxidation of U4+) and of charge balanced ionic substitution as well as defect clustering are discussed. Behavior of urania–yttria is compared to that of corresponding systems in which the tetravalent ion is Ce, Zr, or Hf. The substantial additional stability in the solid solutions compared to pure UO2+x may retard, in both thermodynamic and kinetic sense, the oxidation and leaching of spent fuel to form aqueous U6+ and solid uranyl phases.
Journal of Solid State Chemistry 2008 Volume 181(Issue 1) pp:20-29
Publication Date(Web):January 2008
DOI:10.1016/j.jssc.2007.10.017
Alkali and ammonium cobalt and zinc phosphates show extensive polymorphism. Thermal behavior, relative stabilities, and enthalpies of formation of KCoPO4, RbCoPO4, NH4CoPO4, and NH4ZnPO4 polymorphs are studied by differential scanning calorimetry, high-temperature oxide melt solution calorimetry, and acid solution calorimetry.α-KCoPO4 and γ-KCoPO4 are very similar in enthalpy. γ-KCoPO4 slowly transforms to α-KCoPO4 near 673 K. The high-temperature phase, β-KCoPO4, is 5–7 kJ mol−1 higher in enthalpy than α-KCoPO4 and γ-KCoPO4. HEX phases of NH4CoPO4 and NH4ZnPO4 are about 3 kJ mol−1 lower in enthalpy than the corresponding ABW phases. There is a strong relationship between enthalpy of formation from oxides and acid–base interaction for cobalt and zinc phosphates and also for aluminosilicates with related frameworks. Cobalt and zinc phosphates exhibit similar trends in enthalpies of formation from oxides as aluminosilicates, but their enthalpies of formation from oxides are more exothermic because of their stronger acid–base interactions. Enthalpies of formation from ammonia and oxides of NH4CoPO4 and NH4ZnPO4 are similar, reflecting the similar basicity of CoO and ZnO.Relationship between enthalpy of formation from oxides and acid–base interaction for cobalt phosphates, zinc phosphates, and aluminosilicates with related frameworks. They exhibit similar trends, but the enthalpies of formation of phosphates are more exothermic than those of aluminosilicates because of stronger acid–base interactions.
Iron oxides occur ubiquitously in environmental, geological, planetary, and technological settings. They exist in a rich variety of structures and hydration states. They are commonly fine-grained (nanophase) and poorly crystalline. This review summarizes recently measured thermodynamic data on their formation and surface energies. These data are essential for calculating the thermodynamic stability fields of the various iron oxide and oxyhydroxide phases and understanding their occurrence in natural and anthropogenic environments. The competition between surface enthalpy and the energetics of phase transformation leads to the general conclusion that polymorphs metastable as micrometer-sized or larger crystals can often be thermodynamically stabilized at the nanoscale. Such size-driven crossovers in stability help to explain patterns of occurrence of different iron oxides in nature.
Co-reporter:Qingyuan Liu, Alexandra Navrotsky, Carlos F. Jove-Colon, Francois Bonhomme
Microporous and Mesoporous Materials 2007 Volume 98(1–3) pp:227-233
Publication Date(Web):5 January 2007
DOI:10.1016/j.micromeso.2006.09.008
With its large 12-membered ring channel structure and the periodic arrangement of its ε-cage, cancrinite can host molecular water and a variety of guest anions such as nitrate, carbonate, chloride, and hydroxide. A series of cancrinite samples containing varying amounts of nitrate, carbonate, and water were investigated using high temperature oxide melt drop solution calorimetry. The enthalpies of formation from constituent oxides and from elements at room temperature were obtained. The enthalpy of formation from oxides becomes less exothermic and the water content decreases with increasing salt inclusion (NaNO3 and Na2CO3). This indicates a destabilizing effect of salt hosting on the cancrinite structure. The negative correlation between the amounts of salt and water inclusion suggests competition between water molecules, guest anions and possibly Na cations for the occupancy of the cancrinite channel and cage sites.
Journal of Solid State Chemistry 2007 Volume 180(Issue 9) pp:2443-2451
Publication Date(Web):September 2007
DOI:10.1016/j.jssc.2007.06.017
Differential scanning calorimetry and high temperature oxide melt solution calorimetry were used to study the enthalpy of the α–β phase transformation of NaZnPO4 and enthalpies of formation of α-NaZnPO4, NaH(ZnPO4)2, NaZnPO4·H2O, and NaCoxZn1−xPO4·43H2O (x=0, 0.1, 0.2, 0.3). The enthalpies of formation from the oxides of cobalt substituted in NaZnPO4·43H2O do not depend on cobalt content, confirming similar acid–base interactions for Zn-PO4 and Co-PO4. While water molecules stabilize zinc phosphate frameworks through solvating a cation or forming extra hydrogen bonds, the partial substitution of water for sodium oxide to form NaH(ZnPO4)2 represents the formation of an acidic compound with weaker acid–base interactions and less exothermic enthalpy of formation from oxides than Na2(ZnPO4)2.Relative stability of NaZnPO4 dense phases, open frameworks, and hydrated frameworks. Enthalpy of interaction between water and NaZnPO4 frameworks is presented by reaction: NaZnPO4 (cr, open framework) + nH2O (l) → NaZnPO4·nH2O (cr, hydrated framework).
Co-reporter:Riham M. Morcos, Christoph Mitterbauer, Nigel Browning, Subhash Risbud, Alexandra Navrotsky
Journal of Non-Crystalline Solids 2007 Volume 353(Issue 29) pp:2785-2795
Publication Date(Web):15 September 2007
DOI:10.1016/j.jnoncrysol.2007.05.008
The thermodynamics of CdSe quantum dots embedded in a glass matrix is of great interest because of the numerous applications as optical materials. In this study, the energetics and stability of CdSe quantum dots in a borosilicate glass matrix is investigated as a function of size using high-temperature oxide melt solution calorimetry. CdS0.1Se0.9 nanoparticles (1–40 nm) embedded in glass were analyzed by photoluminescence spectroscopy, electron microprobe, X-ray fluorescence, high-energy synchrotron X-ray diffraction, and (scanning) transmission electron microscopy using both electron energy loss and energy dispersive X-ray spectroscopy. As CdSe particles coarsen, their heat of formation becomes more exothermic. The interfacial energy of CdSe QDs embedded in a borosilicate glass, determined from the slope of enthalpy of drop solution versus calculated surface area, is 0.56 ± 0.01 J/m2.
Co-reporter:M. Dorogova, A. Navrotsky, L.A. Boatner
Journal of Solid State Chemistry 2007 Volume 180(Issue 3) pp:847-851
Publication Date(Web):March 2007
DOI:10.1016/j.jssc.2006.12.001
Rare earth orthovanadates, REVO4, having the zircon structure, form a series of materials interesting for magnetic, optical, sensor, and electronic applications. Enthalpies of formation of REVO4 compounds (RE=Sc, Y, Ce–Nd, Sm–Tm, Lu) were determined by oxide melt solution calorimetry in lead borate (2PbO·2B2O3) solvent at 1075 K. The enthalpies of formation from oxide components become more negative with increasing RE ionic radius. This trend is similar to that obtained for the rare earth phosphates.Comparison of enthalpies of formation from oxides at 298 K for REVO4 [this work] and REPO4 compounds [S.V. Ushakov, K.B. Helean, A. Navrotsky, L.A. Boatner, J. Mater. Res. 16(9) (2001) 2623] vs. RE3+ ionic radius. Filled symbols indicate scheelite structure, open symbols zircon structure.
Co-reporter:Alexandra Navrotsky;Frances Hellman;Maria Dorogova;Charles E. Lesher;Barry L. Zink;David W. Cooke;Juliana Boerio-Goates;Brian Lang;Brian F. Woodfield
PNAS 2007 Volume 104 (Issue 22 ) pp:9187-9191
Publication Date(Web):2007-05-29
DOI:10.1073/pnas.0608165104
Silicon micromachined calorimeters (“calorimeter on a chip”) are used to measure heat capacities and phase transition enthalpies
for thin film, single crystal, and powder samples (5–500 μg). The technology is thus compatible with the small samples produced
in multianvil and large diamond anvil cells. Techniques for handling small samples and attaching them to the calorimetric
devices have been developed. Initial data illustrate application to CoO and to Fe2SiO4 olivine and spinel, a quenched high pressure phase metastable at ambient conditions. The calorimetric entropy of the olivine
- spinel transition in Fe2SiO4 (−16 ± 5 J/mol·K) is in good agreement with that calculated from phase equilibrium data (−14 ± 3 J/mol·K). A magnetic transition
in iron silicate spinel, detected previously by Mossbauer spectroscopy, is seen in the calorimetric signal.
Co-reporter:Eric C. Moloy, Qingyuan Liu, Alexandra Navrotsky
Microporous and Mesoporous Materials 2006 Volume 88(1–3) pp:283-292
Publication Date(Web):21 January 2006
DOI:10.1016/j.micromeso.2005.09.020
The four end-member structures of the hydrosodalite family of materials (Na6+x(OH)x[Al6Si6O24](H2O)N) are synthesized via hydrothermal techniques and characterized using X-ray diffraction, thermal analysis, scanning electron microscopy, and inductively coupled plasma optical emission spectroscopy. High-temperature drop-solution calorimetry in molten 2PbO · B2O3 at 975 K is used to measure the formation and hydration enthalpies. We report the formation enthalpies, from both the oxides and the elements, for a total of eight samples, two for each end-member. The average formation enthalpy (from oxides) is −88.1 ± 1.0 kJ/mol-TO2 for basic sodalite {ideally Na8(OH)2[Al6Si6O24](H2O)4}, −79.6 ± 1.2 kJ/mol-TO2 for hydroxysodalite {ideally Na8(OH)2[Al6Si6O24]}, −68.0 ± 1.1 kJ/mol-TO2, for hydrosodalite {ideally Na6[Al6Si6O24](H2O)8}, and −54.9 ± 1.1 kJ/mol-TO2, for sodalite {ideally Na6[Al6Si6O24]}. The corresponding average hydration enthalpies are −76.7 ± 5.3 kJ/mol-H2O and −37.0 ± 2.4 kJ/mol-H2O for the Na8 and Na6 series, respectively.
Co-reporter:Sanyuan Yang, Qinghua Li, Miaojun Wang, Alexandra Navrotsky
Microporous and Mesoporous Materials 2006 Volume 87(Issue 3) pp:261-267
Publication Date(Web):9 January 2006
DOI:10.1016/j.micromeso.2005.08.016
Eight initially clear solutions (with a general formula based on molar ratios of the components, xNaOH:(5.86 − x)TMAOH:1.0Al2O3:3.40SiO2:370H2O:19.6EtOH:6.0iso-PrOH, x = 0.06–0.86) with various sodium concentrations were prepared for synthesis of FAU/LTA zeolites at 98.0 °C. The synthesis process was studied using in situ calorimetry and the solid products were characterized using XRD, SEM and TG–DSC. At low sodium concentration (x = 0.06), the product is nearly pure FAU while with increasing x from 0.06 to 0.86, LTA gradually becomes dominant. In addition, the sodium concentration has a strong influence on crystal size but limited influence on the chemical composition of the resulting zeolite(s). At x = 0.86, the conversion yields of both silica and alumina are about 50%. When x decreases from 0.86 to 0.06, both conversion yields rapidly decrease to only about 4%. In contrast, the conversion yield of sodium remains at a much higher level (75–100%). There appears to be a maximum crystallization rate at x = 0.16–0.26. Crystallization of both FAU and LTA zeolites is exothermic and the crystallization enthalpy for FAU is higher in magnitude than for LTA.
Co-reporter:Nidhal N. Akl;Olga Trofymluk;Xiubin Qi;Jin Y. Kim;Frank E. Osterloh ;Alexra Navrotsky
Angewandte Chemie International Edition 2006 Volume 45(Issue 22) pp:
Publication Date(Web):26 APR 2006
DOI:10.1002/anie.200503950
Giant holes: Macroporous solids with high electrical conductivity and adjustable pore sizes can be synthesized by the covalent cross-linking of LiMo3Se3 nanowires with citrate-stabilized Ag nanoparticles (see picture, cit=citrate). The ease of synthesis and the possibilities for functionalization with redox-active groups make these xerogels potentially useful in applications such as batteries, capacitors, and electrochemical catalysts.
Journal of Materials Chemistry A 2005 vol. 15(Issue 19) pp:1883-1890
Publication Date(Web):04 Mar 2005
DOI:10.1039/B417143H
Ceramic materials based on the fluorite structure and its derivatives are important for applications in solid oxide fuel cells, sensors, catalysts, gate dielectrics, thermal barrier coatings, and nuclear waste immobilization. The interplay of oxygen vacancy formation and short and long range ordering of vacancies and of cations determines the physical properties and dominates the thermodynamics of these materials. Recent calorimetric data for nanophase zirconia polymorphs, for yttria stabilized zirconia, hafnia, and ceria, and for a series of titanate pyrochlores illustrate the systematics of structure stability relations.
Co-reporter:Qingyuan Liu, Hongwu Xu, Alexandra Navrotsky
Microporous and Mesoporous Materials 2005 Volume 87(Issue 2) pp:146-152
Publication Date(Web):27 December 2005
DOI:10.1016/j.micromeso.2005.08.008
Nitrate cancrinite, with an ideal formula of Na8[Al6Si6O24](NO3)2 · 4H2O, is an important constituent of high-level nuclear wastes at several US Department of Energy (DOE) storage sites. A simple synthesis route for this material is described. Pure nitrate cancrinite has been prepared using a mixed solution of sodium silicate, sodium aluminate, sodium nitrate, and sodium hydroxide. Crystallization took place rapidly at 90 °C and the cancrinite phase emerged within a few hours of aging. Both X-ray diffraction and composition analyses indicated the formation of pure nitrate cancrinite. Solution calorimetric measurements of this material were performed with a Calvet-type calorimeter operated at 701 °C using molten lead borate (2PbO · B2O3) as the solvent. Enthalpies of formation from the constituent oxides and from the elements are −903.3 ± 15.7 kJ/mol and −14,258.3 ± 17.3 kJ/mol, respectively, for a sample with the analyzed formula of Na7.282[Al5.854Si6.146O24](NO3)1.336(CO3)0.046 · 3.365H2O.
Co-reporter:Qinghua Li, Meri L. Amweg, Chanel K. Yee, Alexandra Navrotsky, Atul N. Parikh
Microporous and Mesoporous Materials 2005 Volume 87(Issue 1) pp:45-51
Publication Date(Web):29 December 2005
DOI:10.1016/j.micromeso.2005.07.048
This paper describes an application of a non-thermal, photochemical calcination process for an efficient and spatially controlled removal of the organic structure-directing agent in the preparation of thin films of microporous or zeolite materials. We prepared thin-films of a high silica zeolite (structure code: MFI) following a previously published procedure. The films were illuminated using an ozone generating short-wavelength ultraviolet light in ambient environments and characterized using Fourier-transform infrared spectroscopy, imaging ellipsometry, thin-film X-ray diffraction, and scanning electron microscopy. Results presented here indicate that the UV/ozone treatment under nominally room temperature conditions leads to complete removal of template (structure-directing-agent) from zeolite films comparable to that achieved by thermal calcination. Furthermore, spatially addressing the UV/ozone illumination pattern using a physical mask resulted in the lateral confinement of the template removal from the zeolite film leaving behind a composite film composed of templated and template-free regions. Subsequent chemical treatment of the patterned film selectively removed the as-synthesized, unexposed, regions of the film thereby providing a means for the creation of isolated zeolite film islands at predetermined locations on the substrate surface.
Co-reporter:Sophie Guillemet-Fritsch, Alexandra Navrotsky, Philippe Tailhades, Hervé Coradin, Miaojun Wang
Journal of Solid State Chemistry 2005 Volume 178(Issue 1) pp:106-113
Publication Date(Web):January 2005
DOI:10.1016/j.jssc.2004.10.031
Oxide melt solution calorimetry has been performed on iron manganese oxide spinels prepared at high temperature. The enthalpy of formation of (MnxFe1−x)3O4 at 298 K from the oxides, tetragonal Mn3O4 (hausmannite) and cubic Fe3O4 (magnetite), is negative from x=0x=0 to x=0.67x=0.67 and becomes slightly positive for 0.670.6x>0.6) spinels of intermediate compositions. The enthalpies of formation are discussed in terms of three factors: oxidation–reduction relative to the end-members, cation distribution, and tetragonality. A combination of measured enthalpies and Gibbs free energies of formation in the literature provides entropies of mixing. ΔSmixΔSmix, consistent with a cation distribution in which all trivalent manganese is octahedral and all other ions are randomly distributed for x>0.5x>0.5, but the entropy of mixing appears to be smaller than these predicted values for x<0.4x<0.4.The spinel structure provides octahedral and tetrahedral coordination for cations. In (MnxFe1-x)3O4, strongly negative enthalpies of mixing, measured by oxide melt solution calorimetry, quantify the dominant role of oxidation–reduction reactions (Mn3++Fe2+=Mn2++Fe3+) in the energetics.
Co-reporter:Hongwu Xu, Alexandra Navrotsky, May Nyman, Tina M. Nenoff
Microporous and Mesoporous Materials 2004 Volume 72(1–3) pp:209-218
Publication Date(Web):8 July 2004
DOI:10.1016/j.micromeso.2004.03.033
A complete series of solid solutions with compositions (K1−xCsx)3Ti4Si3O15(OH) · nH2O (n=4–6, 0⩽x⩽1) and having the pharmacosiderite structure (space group P3m) has been synthesized using hydrothermal and ion-exchange methods. Rietveld analysis of synchrotron XRD data shows that the unit-cell parameter a increases linearly with increasing Cs+ content. In the structure, K+ is situated in the center of the eight-membered titanosilicate ring, whereas Cs+ is displaced from the ring center, and the displacement increases with higher K+/(Cs+ + K+) ratio.The enthalpies of formation from the oxides and from the elements were determined by drop solution calorimetry into molten 2PbO · B2O3 solvent at 974 K. The formation enthalpies from oxides become more exothermic with increasing Cs+/(Cs+ + K+), suggesting a stabilizing effect of K+ → Cs+ on the pharmacosiderite structure. Calculation of the enthalpy of the K+ → Cs+ exchange reaction based on the measured formation enthalpies indicates that the Cs+ uptake in these phases is probably thermodynamically (rather than kinetically) driven.
Co-reporter:Atul N. Parikh, Alexandra Navrotsky, Qinghua Li, Chanel K. Yee, Meri L. Amweg, A. Corma
Microporous and Mesoporous Materials 2004 Volume 76(1–3) pp:17-22
Publication Date(Web):1 December 2004
DOI:10.1016/j.micromeso.2004.07.032
We describe a new photochemical method near room temperature conditions for the removal of organic structure-directing agents in the synthesis of microporous materials. The method relies on the exposure of the sample to short-wavelength ultraviolet (UV) radiation in air and the ozone environment generated by a medium pressure mercury lamp (184–257 nm). The generality of the approach has been confirmed using three test-cases of microporous materials: a high-silica synthetic zeolite, an aluminophosphate, and a Ge-substituted microporous silica. The structures and organic contents of the microporous materials before and after UV/ozone treatment were determined using a combination of X-ray diffraction, Fourier-transform infrared spectroscopy, thermogravimetry, and nitrogen adsorption isotherms. For all three cases, the UV/Ozone treatment allows complete removal of the organic template while retaining the inorganic framework. The overall integrity of the microporous materials was comparable to or better than for materials derived by thermal calcination. This method is applicable in making new materials from organic–inorganic precursors and holds promise for microporous thin films on thermally sensitive substrates and for controlled spatial patterning.
Co-reporter:K.B. Helean, S.V. Ushakov, C.E. Brown, A. Navrotsky, J. Lian, R.C. Ewing, J.M. Farmer, L.A. Boatner
Journal of Solid State Chemistry 2004 Volume 177(Issue 6) pp:1858-1866
Publication Date(Web):June 2004
DOI:10.1016/j.jssc.2004.01.009
High-temperature oxide melt solution calorimetry and Rietveld refinements of powder X-ray diffraction data were used to investigate the structure (Fd3m; Z=8) and energetics of a series of RE2Ti2O7 (RE=Sm–Lu) compounds with the pyrochlore structure as well as La2Ti2O7 with a layered perovskite-type structure. All of the RE-titanates were found to be stable in enthalpy with respect to their oxides. In the pyrochlore series, Lu2Ti2O7 was least stable in enthalpy (ΔHf-ox at 298 K=−56.0±4.0 kJ/mol); the most stable materials were Gd-, Eu-, and Sm2Ti2O7 with ΔHf-ox at 298 K=−113.4±2.7, −106.1±4.2, −115.4±4.2 kJ/mol, respectively. In general, as the radius ratio of the A- to B-site cations, RA/RB, decreases, the pyrochlore structure becomes less stable. The trend of ionic radius of the RE3+ cation vs. ΔHf-ox at 298 K is non-linear and approximately parallels the increasing “resistance” to ion-beam-induced amorphization as RA/RB decreases.
Nanoparticle and nanocluster precursors may play a major role in biomineralization. The small differences in enthalpy and
free energy among metastable nanoscale phases offer controlled thermodynamic and mechanistic pathways. Clusters and nanoparticles
offer concentration and controlled transport of reactants. Control of polymorphism, surface energy, and surface charge on
nanoparticles can lead to morphological control and appropriate growth rates of biominerals. Rather than conventional nucleation
and growth, assembly of nanoparticles may provide alternative mechanisms for crystal growth. The Ostwald step rule, based
on a thermodynamic view of nucleation and growth, is supported by the observation that more metastable phases tend to have
lower surface energies. Examples from nonbiological systems, stressing the interplay of thermodynamic and kinetic factors,
illustrate features potentially important to biomineralization.
Co-reporter:Qinghua Li, Alexandra Navrotsky, Fernando Rey, Avelino Corma
Microporous and Mesoporous Materials 2003 Volume 59(2–3) pp:177-183
Publication Date(Web):19 May 2003
DOI:10.1016/S1387-1811(03)00309-3
A series of Ge-zeolites, (GexSi1−x)O2, (ITQ-7, polymorph C and zeolite Beta) with varying Ge content is investigated by high-temperature oxide melt solution calorimetry. The enthalpies of formation relative to a mixture of germania and silica in the quartz structure (ΔHf) are in the range 14.8–21.1 kJ/mol, which implies greater metastability than for pure silica zeolites (6.8–14.4 kJ/mol relative to quartz) and aluminophosphates (5.5–8.5 kJ/mol relative to berlinite on a two-oxygen basis). All the zeolite frameworks examined become energetically less stable with increasing Ge content, but the decrease in stability is considerably different for each framework. The linear regressions of ΔHf vs. Ge content (Ge/(Ge + Si)) for the three structures of Ge-zeolites intersect, though the data are somewhat sparse, implying a possible crossover in framework stability with increasing germanium content. This crossover favors structures with double four member rings (D4MR) at higher Ge/(Ge + Si).
Co-reporter:Qinghua Li, Sanyuan Yang, Alexandra Navrotsky
Microporous and Mesoporous Materials 2003 Volume 65(2–3) pp:137-143
Publication Date(Web):4 November 2003
DOI:10.1016/S1387-1811(03)00480-3
Silicalite-1 nanocrystals were synthesized with different sizes (40, 95 and 180 nm) from clear solutions and the organic structure directing agent removed by calcination. X-ray diffraction, scanning electron microscopy, nitrogen adsorption, Fourier transform infrared spectroscopy and thermogravimetric analysis were used for characterization. High-temperature solution calorimetry using lead borate (2PbO · B2O3) solvent at 974 K measured the enthalpies (ΔHtran) relative to quartz at 298 K. The ΔHtran values are 8.2 ± 0.4 kJ/mol (40 nm), 8.3 ± 0.3 kJ/mol (95 nm) and 8.0 ± 0.5 kJ/mol (180 nm). These values are essentially the same as ΔHtran for microsized MFI crystals, 8.0 ± 0.8 kJ/mol [J. Phys. Chem. B 104 (2000) 10001]. Thus the particle size has no effect on the framework energetics of silicalite. This unusual phenomenon, which is different from that of condensed nanocrystalline metal oxides, is discussed in terms of internal surface area and surface energy of zeolites.
Co-reporter:Lan Wang, Alexandra Navrotsky, Rebecca Stevens, Brian F. Woodfield, Juliana Boerio-Goates
The Journal of Chemical Thermodynamics 2003 Volume 35(Issue 7) pp:1151-1159
Publication Date(Web):July 2003
DOI:10.1016/S0021-9614(03)00083-1
Enthalpies of mixing in CoxMg1 − xO solid solutions at T=298 K have been determined using high temperature drop solution calorimetry in molten sodium molybdate (3Na2O · 4MoO3) solvent at T=973 K. Slightly positive enthalpies of mixing were observed, which conform to a regular solution model with interaction parameter W=(5.1±0.3) kJ · mol−1, which is similar to the interaction parameter calculated from previously measured activity-composition relations. Standard molar entropies Sm0 at T=298 K calculated from subambient Cp data for CoO, MgO are (52.8 ± 0.1) J · K−1 · mol−1 and (28.1 ± 0.1) J · K−1 · mol−1, respectively. Integration of the Cp,m/T of the Co0.50Mg0.50O mixture yields an entropy of (40.3 ± 0.1) J · K−1 · mol−1. This value does not include configurational entropy from the Co/Mg site disorder. The excess entropy of Co0.50Mg0.50O is zero within experimental error. Heat capacities were also measured between T=298 K and T=973 K. The enthalpies of mixing do not depend on temperature in this range.
Co-reporter:Eric C. Moloy, Lilian P. Davila, James F. Shackelford, Alexandra Navrotsky
Microporous and Mesoporous Materials 2002 Volume 54(1–2) pp:1-13
Publication Date(Web):1 July 2002
DOI:10.1016/S1387-1811(02)00328-1
High-silica zeolites are 5.6–15.5 kJ/mol less stable in enthalpy than α-quartz––the stable polymorph of silica under ambient conditions. Previous studies have correlated these energetic metastabilities to molar volumes and framework densities. In this study, we consider the question of whether these energetics might arise from a surface free energy term that originates from the large internal surfaces of these materials. Cerius2 molecular simulation software is used to calculate the internal surface areas. A linear relationship between formation enthalpy and internal surface area is found for α-quartz, α-cristobalite, and 17 zeolitic frameworks: AFI, AST, BEA, CFI, CHA, EMT, FAU, FER, IFR, ISV, ITE, MEI, MEL, MFI, MTW, MWW, and STT. The slope of the regression line has direct physical meaning: an average internal surface enthalpy of 0.093±0.009 J/m2. This value is similar to a value of 0.100±0.035 J/m2 for the average external surface free energy of amorphous silica obtained from various amorphous, but not microporous or mesoporous, phases reported in the literature. We conclude that it is physically reasonable to consider the metastability of anhydrous silica zeolites as resulting from their large internal surface area, that the average value of the surface enthalpy (or surface free energy) is similar for both internal and external surfaces, and that this quantity is not strongly dependent on the specific nature of the tetrahedral framework.
The energetics of the TiO2 polymorphs (rutile, anatase, and brookite) were studied by high temperature oxide melt drop solution calorimetry. Relative
to bulk rutile, bulk brookite is 0.71 ± 0.38 kJ/mol (6) and bulk anatase is 2.61 ± 0.41 kJ/mol higher in enthalpy. The surface
enthalpies of rutile, brookite, and anatase are 2.2 ± 0.2 J/m2, 1.0 ± 0.2 J/m2, and 0.4 ± 0.1 J/m2, respectively. The closely balanced energetics directly confirm the crossover in stability of nanophase polymorphs inferred
by Zhang and Banfield (7). An amorphous sample with surface area of 34,600 m2/mol is 24.25 ± 0.88 kJ/mol higher in enthalpy than bulk rutile.
Co-reporter:Sanyuan Yang, Alexandra Navrotsky, Brian L Phillips
Microporous and Mesoporous Materials 2001 Volume 46(2–3) pp:137-151
Publication Date(Web):August 2001
DOI:10.1016/S1387-1811(01)00268-2
Synthesis of FAU zeolite from a mixture of silicate and aluminate solutions (5.15Na2O–1.00Al2O3–3.28SiO2–165H2O) was studied using an in situ calorimetric method at a heating rate of 0.1°C/min. The scanning calorimetric curve provides a coherent and consistent recording of the synthesis process. At low temperature (25–66°C), the calorimetric curve drifts slightly in the endothermic direction. Chemical analysis shows a slight increase in the dissolution of the amorphous gel/solid phase with increasing temperature in this period. The onset of the exothermic peak at 66.7°C is suggested to signal the beginning of the nucleation/crystallization process. Formation of the FAU structure is an exothermic event and the associated heat effect was directly measured, −2.38±0.06 kJ/mol based on TO2 (T=Si or Al) or −457±12 kJ/mol based on the unit cell formula of the FAU product, Na91 · Si105Al91O384 · 277H2O. The integral heat for the crystallization by calorimetry agrees well with the X-ray diffraction (XRD) or NMR crystallinity of the solid phase. At the early stages of crystallization (integral heat <0.1% of the total) the compositional changes in solid and solution were similar to those in the pre-crystallization period. The FAU phase was slightly richer in Al than the gel/solid amorphous precursor. The abrupt drop of the soluble Al concentration during the rapid crystallization period indicated the direct participation of the soluble aluminosilicate species in the crystal growth. The crystalline phase could be observed using XRD, NMR or IR only significantly after the onset of the calorimetric peak because the mass fraction of the nuclei/crystals in the solid phase was initially too low to be detected by the former methods. This demonstrated the high sensitivity of the calorimetric method.
Co-reporter:Hongwu Xu, Yiping Zhang, Alexandra Navrotsky
Microporous and Mesoporous Materials 2001 Volume 47(2–3) pp:285-291
Publication Date(Web):October 2001
DOI:10.1016/S1387-1811(01)00388-2
The energetics of microporous titanosilicates ETS-4 (K1.13Na3.92Ti3.07Si8.17O25·8.64H2O) and ETS-10 (K0.61Na1.09Ti1.10Si4.98O13·2.89H2O) has been investigated by high-temperature drop solution calorimetry using lead borate as the solvent at 974 K. Combining the measured heats of drop solution with the published enthalpies of drop solution and formation for the constituent oxides, the standard enthalpies of formation from the oxides (ΔH0f,ox) and from the elements (ΔHf,el0) for both phases were derived for the first time. The obtained values (in kJ/mol) are as follows: ΔHf,ox0(ETS-4)=−818.5±13.1, ΔHf,el0(ETS-4)=−14,642.8±15.9, ΔHf,ox0(ETS-10)=−262.2±3.1, and ΔHf,el0(ETS-10)=−6995.2±6.1. Comparison between the ΔHf,ox0 (or ΔHf,el0) values of the two phases suggests that ETS-4 is thermodynamically more stable than ETS-10 with respect to the oxides (or the elements) at 298 K and 1 atm. This behavior can largely be attributed to the higher degree of hydration of ETS-4 than that of ETS-10.
Co-reporter:Martin C. Wilding, Alexandra Navrotsky
Journal of Non-Crystalline Solids 2000 Volume 265(Issue 3) pp:238-251
Publication Date(Web):March 2000
DOI:10.1016/S0022-3093(00)00007-7
High temperature calorimetry at 1760 K has been used to measure the heat of solution of La2O3 in a series of simple alkali and alkaline earth silicate liquids. The heat of solution in these solvents is strongly exothermic and varies as a function of liquid composition. However, the variation of the heat of solution does not follow simple trends related to cation size or charge and varies little with La2O3 concentration. The variation of heat of solution with composition of the liquid reflects the ability of La(III) to perturb the transient silicate framework and compete with other cations for oxygen. This complex pattern of melt energetics is consistent with recent spectroscopic measurements which suggest extreme perturbation of the silicate framework by La(III), sufficient to isolate oxygen from silicon. This interpretation suggests the presence of phase-ordered regions rich in La(III) consistent with incipient liquid–liquid immiscibility suggested by previous calorimetric studies. Within error the heat capacity of La-bearing silicate liquids is the same over the super-cooled liquid range as in the stable liquid, with no evidence for the large heat capacities associated with melt restructuring. Thus the energetics of the liquid are dominated by the exothermic reactions which form La-clusters and these phase-ordered regions do not dissociate as temperature increases up to 1760 K.
Co-reporter:Irina Molodetsky, Alexandra Navrotsky, Michael J. Paskowitz, Valerie J. Leppert, Subhash H. Risbud
Journal of Non-Crystalline Solids 2000 Volume 262(1–3) pp:106-113
Publication Date(Web):February 2000
DOI:10.1016/S0022-3093(99)00690-0
X-ray-amorphous zirconia was synthesized by a low temperature process. The difference in the energetics of monoclinic and amorphous zirconia was calculated based on drop solution calorimetry of amorphous and monoclinic zirconia in lead borate (2PbO·B2O3) at 1073 K. X-ray-amorphous zirconia is about 58 kJ/mol less stable in enthalpy than the monoclinic phase. Amorphous zirconia heated to 573 K shows crystalline nanoparticles (cubic or tetragonal) of 3.8 nm average diameter. Formation of amorphous zirconia at low temperatures can be explained by its surface energy stabilization relative to monoclinic zirconia, with a difference .
Co-reporter:Naiwang Liu, Xiaofeng Guo, Alexandra Navrotsky, Li Shi, Di Wu
Journal of Catalysis (October 2016) Volume 342() pp:158-163
Publication Date(Web):1 October 2016
DOI:10.1016/j.jcat.2016.08.001
•A series of sulfated zirconia catalysts were synthesized and characterized.•Thermochemical insights into sulfated zirconia synthesis were revealed.•Enthalpies of sulfated zirconia formation were probed directly using calorimetry.•Energetic trend of sulfated zirconia catalysts was experimentally determined.•Thermodynamics may provide crucial insights into catalytic material development.A series of sulfated zirconia (SZ) catalysts were synthesized by immersion of amorphous zirconium hydroxide in sulfuric acid of various concentrations (1–5 N). These samples were fully characterized by X-ray diffraction (XRD), thermogravimetric analysis and mass spectrometry (TGA-MS), and aqueous sulfuric acid immersion and high temperature oxide melt solution calorimetry. We investigated the enthalpies of the complex interactions between sulfur species and the zirconia surface (ΔHSZ) for the sulfated zirconia precursor (SZP), ranging from −109.46 ± 7.33 (1 N) to −42.50 ± 0.89 (4 N) kJ/mol S. ΔHSZ appears to be a roughly exponential function of sulfuric acid concentration. On the other hand, the enthalpy of SZ formation (ΔHf), becomes more exothermic linearly as sulfur surface coverage increases, from −147.90 ± 4.16 (2.14 nm−2) to −317.03 ± 4.20 (2.29 nm−2) kJ/mol S, indicating formation of energetically more stable polysulfate species.Download high-res image (44KB)Download full-size image
Co-reporter:Tatiana Y. Shvareva, Lena Mazeina, Drew Gorman-Lewis, Peter C. Burns, Jennifer E.S. Szymanowski, Jeremy B. Fein, Alexandra Navrotsky
Geochimica et Cosmochimica Acta (15 September 2011) Volume 75(Issue 18) pp:5269-5282
Publication Date(Web):15 September 2011
DOI:10.1016/j.gca.2011.06.041
Boltwoodite and uranophane are uranyl silicates common in oxidized zones of uranium ore deposits. An understanding of processes that impact uranium transport in the environment, especially pertaining to the distribution of uranium between solid phases and aqueous solutions, ultimately requires determination of thermodynamic parameters for such crystalline materials. We measured formation enthalpies of synthetic boltwoodites, K(UO2)(HSiO4)·H2O and Na(UO2)(HSiO4)·H2O, and uranophane, Ca(UO2)2(HSiO4)2·5H2O, by high temperature oxide melt solution calorimetry. We also studied the aqueous solubility of these phases from both saturated and undersaturated conditions at a variety of pH. The combined data permit the determination of standard enthalpies, entropies and Gibbs free energies of formation for each phase and analysis of its potential geological impact from a thermodynamic point of view.
Co-reporter:Yannick Linard, Martin C. Wilding, Alexandra Navrotsky
Geochimica et Cosmochimica Acta (15 January 2008) Volume 72(Issue 2) pp:590-601
Publication Date(Web):15 January 2008
DOI:10.1016/j.gca.2007.10.004
The enthalpies of solution of La2O3, TiO2, HfO2, NiO and CuO were measured in sodium silicate melts at high temperature. When the heat of fusion was available, we derived the corresponding liquid–liquid enthalpies of mixing. These data, combined with previously published work, provide insight into the speciation reactions in sodium silicate melts. The heat of solution of La2O3 in these silicate solvents is strongly exothermic and varies little with La2O3 concentration. The variation of heat of solution with composition of the liquid reflects the ability of La(III) to perturb the transient silicate framework and compete with other cations for oxygen. The enthalpy of solution of TiO2 is temperature-dependent and indicates that the formation of Na–O–Si species is favored over Na–O–Ti at low temperature. The speciation reactions can be interpreted in terms of recent spectroscopic studies of titanium-bearing melts which identify a dual role of Ti4+ as both a network-former end network-modifier. The heats of solution of oxides of transition elements (Ni and Cu) are endothermic, concentration-dependent and reach a maximum with concentration. These indicate a charge balanced substitution which diminishes the network modifying role of Na+ by addition of Ni2+ or Cu2+. The transition metal is believed to be in tetrahedral coordination, charge balanced by the sodium cation in the melts.
Co-reporter:Ferenc Lázár Forray, A.M.L. Smith, A. Navrotsky, K. Wright, K.A. Hudson-Edwards, W.E. Dubbin
Geochimica et Cosmochimica Acta (15 February 2014) Volume 127() pp:107-119
Publication Date(Web):15 February 2014
DOI:10.1016/j.gca.2013.10.043
The enthalpy of formation from the elements of well characterized Pb–As, Pb–Cu, and Pb–Zn synthetic jarosites, corresponding to chemical formulas (H3O)0.68±0.03Pb0.32±0.002Fe2.86±0.14(SO4)1.69±0.08(AsO4)0.31±0.02(OH)5.59±0.28(H2O)0.41±0.02, (H3O)0.67±0.03Pb0.33±0.02Fe2.71±0.14Cu0.25±0.01(SO4)2±0.00(OH)5.96±0.30(H2O)0.04±0.002 and (H3O)0.57±0.03Pb0.43±0.02Fe2.70±0.14Zn0.21±0.01(SO4)2±0.00(OH)5.95±0.30(H2O)0.05±0.002, was measured by high temperature oxide melt solution calorimetry and gave ΔH°f = −3691.2 ± 8.6 kJ/mol, ΔH°f = −3653.6 ± 8.2 kJ/mol, and ΔH°f = −3669.4 ± 8.4 kJ/mol, respectively. Using estimated entropies, the standard Gibbs free energy of formation from elements at 298 K ΔG°f of the three compounds were calculated to be −3164.8 ± 9.1, −3131.4 ± 8.7, and −3153.6 ± 8.9 kJ/mol, respectively. Based on these free energies, their log Ksp values are −13.94 ± 1.89, −4.38 ± 1.81 and −3.75 ± 1.80, respectively. For this compounds, a log10{Pb2+}–pH diagram is presented. The diagram shows that the formation of Pb–As jarosite may decrease aqueous arsenic and lead concentrations to meet drinking water standards. The new thermodynamic data confirm that transformation of Pb–As jarosite to plumbojarosite is thermodynamically possible.
Co-reporter:Lena Mazeina, Alexandra Navrotsky, Darby Dyar
Geochimica et Cosmochimica Acta (15 February 2008) Volume 72(Issue 4) pp:1143-1153
Publication Date(Web):15 February 2008
DOI:10.1016/j.gca.2007.11.032
As a contribution to the systematic study of iron oxide thermodynamics, this work reports enthalpies of formation of green rust, a double layered (FeII, FeIII) hydroxide with the ideal stoichiometry FeII1-xFeIIIx(OH)2[Am-]y/m·nH2O, with sulfate as the anion in the interlayer. Samples were characterized by X-ray powder diffraction, thermogravimetric analysis, infrared spectroscopy, and Mössbauer spectroscopy. Full chemical analysis was performed. Contents of FeII, FeIII, water, and sulfate were obtained. We report standard enthalpies of formation for green rust with different FeII/FeIII ratios. Enthalpies of formation from single cation compounds, namely, Fe(OH)2, Fe(OH)3, FeSO4 and H2O show reasonable agreement with Gibbs free energies of formation from single cation compounds recalculated from the reported literature values. These values show that green rust has little stabilization over a mechanical mixture of these single cation compounds and there is no thermodynamic preference for any particular FeII/FeIII ratio.
Co-reporter:Tori Z. Forbes, A.V. Radha, Alexandra Navrotsky
Geochimica et Cosmochimica Acta (15 December 2011) Volume 75(Issue 24) pp:7893-7905
Publication Date(Web):15 December 2011
DOI:10.1016/j.gca.2011.09.034
Calcium carbonate (CaCO3) is an important component of the near-surface environment. Understanding the nature of its precipitation is important for a variety of environmental processes, as well as for the geologic sequestration of anthropogenic carbon dioxide. Calcite is the most thermodynamically stable bulk polymorph, but energy crossovers may exist that could favor the precipitation of vaterite or aragonite with decreasing particle size. The purpose of this study is to determine the surface energy of calcite, which is the first step towards understanding the effect of particle size on thermodynamic stability in the calcium carbonate system. The enthalpies of five well-characterized calcite samples (four nanophase and one bulk) were measured by acid solution isothermal and water adsorption calorimetric techniques. From the calorimetric data, the surface energies of calcite were determined to be 1.48 ± 0.21 and 1.87 ± 0.16 J/m2 for hydrous and anhydrous surfaces. These values are similar to those measured for many oxides but larger than predicted from computational models for idealized calcite surfaces. The surfaces of synthetic CaCO3 particles contain a range of planes and defect structures, which may give rise to the difference between the experimental and modeled values.
Geochimica et Cosmochimica Acta (15 April 2007) Volume 71(Issue 8) pp:2072-2078
Publication Date(Web):15 April 2007
DOI:10.1016/j.gca.2007.01.011
The formation of nitrate sodalite, an important constituent of the resilient heels at DOE nuclear waste storage sites, was closely followed by oven synthesis, in situ calorimetry as a function of heating rate from 0.01 to 0.1 °C/min and X-ray diffraction. A transition sequence of amorphous–zeolite A–sodalite–cancrinite was confirmed. For in situ synthesis calorimetry, the heat flow peaks related to zeolite A formation are shifted to higher temperatures as heating rate increases. Although the end products are mostly nitrate sodalite, no calorimetric signals associated with zeolite A to sodalite conversion are detected. This suggests that the enthalpy of formation of zeolite A and sodalite are very similar and the zeolite A to sodalite conversion enthalpy is small. This conclusion is in accord with previous measurements by oxide melt solution calorimetry.
Co-reporter:F.L. Forray, A.M.L. Smith, C. Drouet, A. Navrotsky, K. Wright, K.A. Hudson-Edwards, W.E. Dubbin
Geochimica et Cosmochimica Acta (1 January 2010) Volume 74(Issue 1) pp:215-224
Publication Date(Web):1 January 2010
DOI:10.1016/j.gca.2009.09.033
The enthalpy of formation from the elements of a well-characterized synthetic Pb-jarosite sample corresponding to the chemical formula (H3O)0.74Pb0.13Fe2.92(SO4)2(OH)5.76(H2O)0.24 was measured by high temperature oxide melt solution calorimetry. This value (ΔH∘f = −3695.9 ± 9.7 kJ/mol) is the first direct measurement of the heat of formation for a lead-containing jarosite. Comparison to the thermochemical properties of hydronium jarosite and plumbojarosite end-members strongly suggests the existence of a negative enthalpy of mixing possibly related to the nonrandom distribution of Pb2+ ions within the jarosite structure. Based on these considerations, the following thermodynamic data are proposed as the recommended values for the enthalpy of formation from the elements of the ideal stoichiometric plumbojarosite Pb0.5Fe3(SO4)2(OH)6: ΔG∘f = −3118.1 ± 4.6 kJ/mol, ΔH∘f = −3603.6 ± 4.6 kJ/mol and S° = 376.6 ± 4.5 J/(mol K). These data should prove helpful for the calculation of phase diagrams of the Pb–Fe–SO4–H2O system and for estimating the solubility product of pure plumbojarosite. For illustration, the evolution of the estimated solubility product of ideal plumbojarosite as a function of temperature in the range 5–45 °C was computed (Log(Ksp) ranging from −24.3 to −26.2). An Eh–pH diagram is also presented.
Co-reporter:D. Kapush, S.V. Ushakov, A. Navrotsky, Q.-J. Hong, H. Liu, A. van de Walle
Acta Materialia (1 February 2017) Volume 124() pp:
Publication Date(Web):1 February 2017
DOI:10.1016/j.actamat.2016.11.003
“Drop-n-catch” calorimetry was performed on Y2O3 spheroids of 2–3 mm in diameter, prepared by laser melting of powders. Samples were aerodynamically levitated in a splittable nozzle levitator in air or argon flow, laser heated from ∼2200 to 3000 °C and dropped into a calorimeter at 25 °C, thus measuring their enthalpy as a function of temperature. The fusion enthalpy of cubic Y2O3 was derived from the step in the temperature–enthalpy curve as 119 ± 10 kJ/mol. Calculations performed using density functional theory and molecular dynamics techniques produced the value 127 ± 3 kJ/mol Y2O3. This combined methodology enables accurate determination of the enthalpies of fusion and phase transition of refractory oxides, including those containing lanthanides and actinides.
Co-reporter:Xiaofeng Guo, Christian Lipp, Eitan Tiferet, Antonio Lanzirotti, Matthew Newville, Mark H. Engelhard, Di Wu, Eugene S. Ilton, Stephen R. Sutton, Hongwu Xu, Peter C. Burns and Alexandra Navrotsky
Heating a mixture of uranyl(VI) nitrate and tantalum(V) oxide in the molar ratio of 2:3 to 1400 °C resulted in the formation of a new compound, UTa3O10. The honey colored to yellow brown crystals of UTa3O10 crystallize in an orthorhombic structure with the space group Fddd (no. 70), lattice parameters a = 7.3947(1), b = 12.7599(2), c = 15.8156(2) Å, and Z = 8. Vertex sharing [TaO6]7− octahedra of two crystallographically distinct Ta cations form a three dimensional tantalate framework. Within this framework, six membered rings of [TaO6]7− octahedra are formed within the (001) plane. The center of these rings is occupied by the uranyl cations [UO2]+, with an oxidation state of +5 for uranium. The pentavalence of U and Ta was confirmed by X-ray photoelectron spectroscopy and X-ray adsorption spectroscopy. The enthalpy of formation of UTa3O10 from Ta2O5, β-U3O7, and U3O8 has been determined to be 13.1 ± 18.1 kJ mol−1 using high temperature oxide melt solution calorimetry with sodium molybdate as the solvent at 700 °C. The close to zero enthalpy of formation of UTa3O10 can be explained by closely balanced structural stabilizing and destabilizing factors, which may also apply to other UM3O10 compounds.
Co-reporter:Sriram Goverapet Srinivasan, Radha Shivaramaiah, Paul R. C. Kent, Andrew G. Stack, Richard Riman, Andre Anderko, Alexandra Navrotsky and Vyacheslav S. Bryantsev
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 11) pp:NaN7832-7832
Publication Date(Web):2017/02/24
DOI:10.1039/C7CP00811B
Bastnäsite, a fluoro-carbonate mineral, is the single largest mineral source of light rare earth elements (REE), La, Ce and Nd. Enhancing the efficiency of separation of the mineral from gangue through froth flotation is the first step towards meeting an ever increasing demand for REE. To design and evaluate collector molecules that selectively bind to bastnäsite, a fundamental understanding of the structure and surface properties of bastnäsite is essential. In our earlier work (J. Phys. Chem. C, 2016, 120, 16767), we carried out an extensive study of the structure, surface stability and water adsorption energies of La-bastnäsite. In this work, we make a comparative study of the surface properties of Ce-bastnäsite, La-bastnäsite, and calcite using a combination of density functional theory (DFT) and water adsorption calorimetry. Spin polarized DFT+U calculations show that the exchange interaction between the electrons in Ce 4f orbitals is negligible and that these orbitals do not participate in bonding with the oxygen atom of the adsorbed water molecule. In agreement with calorimetry, DFT calculations predict larger surface energies and stronger water adsorption energies on Ce-bastnäsite than on La-bastnäsite. The order of stabilities for stoichiometric surfaces is as follows: [100] > [101] > [102] > [0001] > [112] > [104] and the most favorable adsorption sites for water molecules are the same as for La-bastnäsite. In agreement with water adsorption calorimetry, at low coverage water molecules are strongly stabilized via coordination to the surface Ce3+ ions, whereas at higher coverage they are adsorbed less strongly via hydrogen bonding interaction with the surface anions. Due to similar water adsorption energies on bastnäsite [101] and calcite [104] surfaces, the design of collector molecules that selectively bind to bastnäsite over calcite must exploit the structural differences in the predominantly exposed facets of these minerals.
Co-reporter:Xin Guo, Pardha Saradhi Maram and Alexandra Navrotsky
Journal of Materials Chemistry A 2017 - vol. 5(Issue 25) pp:NaN12957-12957
Publication Date(Web):2017/05/17
DOI:10.1039/C7TA02434G
Perovskite-structured lithium lanthanum titanate (LLTO) Li3xLa0.67−xTiO3 (compositions x = 0.04 to 0.15) has been prepared by conventional solid state reaction. The phase purity and crystal structural changes were investigated with XRD, FTIR and Raman. The vibrational spectra reveal the interaction between metal cation and oxygen anion with increasing Li doping and structural evolution. LLTO and component oxides were studied by high temperature oxide melt solution calorimetry. The formation enthalpies of LLTO from oxides are exothermic for all compositions, indicating thermodynamic stability. There are two regimes in the trend of formation enthalpy with increasing Li concentration. In the first regime, x ≤0.08, the formation enthalpies vary slowly with composition, but the lowest stability by about 1.5 kJ mol−1 is seen at x = 0.06. An abrupt change in the formation enthalpy trend is observed in the second regime when x ≥0.1, where maximum lithium ion conductivity (at x = 0.10) is reported. The least stable composition, x = 0.06, occurs where maximum charge carrier concentration and lowest activation energy is reported. From the thermodynamic study, it is clear that the energetically least stable composition correlates with lowest activation energy whereas the sharp change in formation enthalpy trend correlates with highest Li-ion conductivity.
Co-reporter:Benjamin E. Hanken, Tatiana Y. Shvareva, Niels Grønbech-Jensen, Christopher R. Stanek, Mark Asta and Alexandra Navrotsky
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 16) pp:NaN5685-5685
Publication Date(Web):2012/03/05
DOI:10.1039/C2CP40295E
Cation mixing energetics in urania–ceria solid solutions with stoichiometric oxygen concentrations (U1−yCeyO2) have been measured by high-temperature oxide-melt drop-solution calorimetry. Measurements have been performed on eight samples with compositions spanning y = 0.119 to y = 0.815. The measured mixing enthalpies (ΔHmix) range from −0.6 ± 3.3 to 3.9 ± 3.0 kJ mol−1. These values are discussed in the context of results from atomistic modeling which take into consideration the possibility of charge transfer between uranium and cerium cations to form solid solutions with mixed charge states. A comparison between measured and calculated results for ΔHmix suggests that such charge transfer takes place to a limited extent in the most concentrated mixtures studied.
Co-reporter:Sulata K. Sahu, Baiyu Huang, Kristina Lilova, Brian F. Woodfield and Alexandra Navrotsky
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 34) pp:NaN22295-22295
Publication Date(Web):2015/07/22
DOI:10.1039/C5CP02972D
High temperature oxide melt solution calorimetry has been performed to investigate the enthalpies of mixing (ΔmixH) of bulk and nanophase (1 − x)Fe3O4–xM3O4 (M = Co, Mn) spinel solid solutions. The entropies of mixing (ΔmixS) were calculated from the configurational entropies based on cation distributions, and the Gibbs free energies of mixing (ΔmixG) were obtained. The ΔmixH and ΔmixG for the (1 − x)Fe3O4–xCo3O4 system are negative over the complete solid solution range, for both macroscopic and nanoparticulate materials. In (1 − x)Fe3O4–xMn3O4, the formation enthalpies of cubic Fe3O4 (magnetite) and tetragonal Mn3O4 (hausmannite) are negative for Mn3O4 mole fractions less than 0.67 and slightly positive for higher manganese content. Relative to cubic Fe3O4 and cubic Mn3O4 (stable at high temperature), the enthalpies and Gibbs energies of mixing are negative over the entire composition range. A combination of measured mixing enthalpies and reported Gibbs energies in the literature provides experimental entropies of mixing. The experimental entropies of mixing are consistent with those calculated from cation distributions for x > 0.3 but are smaller than those predicted for x < 0.3. This discrepancy may be related to the calculations, having treated Fe2+ and Fe3+ as distinguishable species. The measured surface energies of the (1 − x)Fe3O4–xM3O4 solid solutions are in the range of 0.6–0.9 J m−2, similar to those of many other spinels. Because the surface energies are relatively constant, the thermodynamics of mixing at a given particle size throughout the solid solution can be considered independent of the particular particle size, thus confirming and extending the conclusions of a recent study on iron spinels.
Co-reporter:H. Sun, D. Wu, X. Guo, B. Shen and A. Navrotsky
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 17) pp:NaN11203-11203
Publication Date(Web):2015/03/25
DOI:10.1039/C5CP01133G
A series of calcium-exchanged zeolite A samples with different degrees of exchange were prepared. They were characterized by powder X-ray diffraction (XRD) and differential scanning calorimetry (DSC). High temperature oxide melt drop solution calorimetry measured the formation enthalpies of hydrated zeolites CaNa-A from constituent oxides. The water content is a linear function of the degree of exchange, ranging from 20.54% for Na-A to 23.77% for 97.9% CaNa-A. The enthalpies of formation (from oxides) at 25 °C are −74.50 ± 1.21 kJ mol−1 TO2 for hydrated zeolite Na-A and −30.79 ± 1.64 kJ mol−1 TO2 for hydrated zeolite 97.9% CaNa-A. Dehydration enthalpies obtained from differential scanning calorimetry are 32.0 kJ mol−1 H2O for hydrated zeolite Na-A and 20.5 kJ mol−1 H2O for hydrated zeolite 97.9% CaNa-A. Enthalpies of formation of Ca-exchanged zeolites A are less exothermic than for zeolite Na-A. A linear relationship between the formation enthalpy and the extent of calcium substitution was observed. The energetic effect of Ca-exchange on zeolite A is discussed with an emphasis on the complex interactions between the zeolite framework, cations, and water.
Co-reporter:H. Sun, D. Wu, X. Guo and A. Navrotsky
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 14) pp:NaN9247-9247
Publication Date(Web):2015/02/26
DOI:10.1039/C5CP00016E
The properties of zeolite A change significantly upon sodium–calcium exchange. The impact of cation composition on the temperature-induced phase transformations and energetics of Na–Ca exchanged zeolite A was studied systematically using powder X-ray diffraction (XRD), thermogravimetric analysis (TGA), differential scanning calorimetry (DSC) and high-temperature oxide melt solution calorimetry. As the temperature increases, the structural evolution of each Na–Ca exchanged zeolite A sample undergoes three distinct stages – dehydration, amorphization, and densification/recrystallization. Initially complete dehydration does not result in framework degradation, but further heating leads to zeolite phase degradation into other aluminosilicate phases. Both amorphization and recrystallization shift to higher temperatures as the calcium content increases. On the other hand, the enthalpies of formation for the high temperature aluminosilicate phases, the amorphous phase (AP) and the dense phase (DP), appear to be a linear function of calcium content (average ionic potential) with diminishing of energetic stability upon increasing the Ca content. 100% Na-A heated at 1200 °C has the most exothermic enthalpy of formation from oxides (−65.87 ± 0.87 kJ mol−1 – TO2), while 97.9% CaNa-A heated at 945 °C has the least exothermic value (−5.26 ± 0.62 kJ mol−1 – TO2). For different aluminosilicates with the same chemical composition, the dense phase (DP) assemblage is more stable than the amorphous phase (AP).
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 15) pp:NaN10122-10122
Publication Date(Web):2016/03/10
DOI:10.1039/C5CP07918G
Transition metal cations (Mn2+, Co2+, Cu2+, and Zn2+) containing zeolites A and Y were synthesized by ion exchange and their thermochemistry was investigated by differential scanning calorimetry and high temperature oxide melt solution calorimetry. The enthalpies of formation from oxides for Mn, Co, Cu, and Zn zeolites A range from 14.0 ± 1.3 to 67.6 ± 5.5 kJ mol−1 and those for Mn, Co, Cu, and Zn zeolites Y range from 8.0 ± 2.0 to 32.6 ± 1.8 kJ mol−1. All these zeolites are thus metastable with respect to oxide components and to other dense phases. The formation enthalpies of Mn and Zn exchanged zeolites A and Y are less endothermic than those of corresponding Co and Cu exchanged A and Y. These energetics are consistent with metal oxygen bond lengths and related to crystal field effects of transition metal ions. Similar thermodynamic trends have been seen in transition metal containing spinel, olivine and pyroxene materials. The enthalpies of exchange of transition metals in zeolites A and Y with sodium in aqueous solution are calculated and suggest that these zeolites could be reasonably effective sorbents for heavy metal waste.
Co-reporter:Zamirbek Akimbekov, Di Wu, Carl K. Brozek, Mircea Dincă and Alexandra Navrotsky
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 2) pp:NaN1162-1162
Publication Date(Web):2015/11/26
DOI:10.1039/C5CP05370F
The inclusion of solvent in metal–organic framework (MOF) materials is a highly specific form of guest–host interaction. In this work, the energetics of solvent MOF-5 interactions has been investigated by solution calorimetry in 5 M sodium hydroxide (NaOH) at room temperature. Solution calorimetric measurement of enthalpy of formation (ΔHf) of Zn4O(C8H4O4)3·C3H7NO (MOF-5·DMF) and Zn4O(C8H4O4)3·0.60C5H11NO (MOF-5·0.60DEF) from the dense components zinc oxide (ZnO), 1,4-benzenedicarboxylic acid (H2BDC), N,N-dimethylformamide (DMF) and N,N-diethylformamide (DEF) gives values of 16.69 ± 1.21 and 45.90 ± 1.46 kJ (mol Zn4O)−1, respectively. The enthalpies of interaction (ΔHint) for DMF and DEF with MOF-5 are −82.78 ± 4.84 kJ (mol DMF)−1 and −89.28 ± 3.05 kJ (mol DEF)−1, respectively. These exothermic interaction energies suggest that, at low guest loading, Lewis base solvents interact more strongly with electron accepting Zn4O clusters in the MOF than at high solvent loading. These data provide a quantitative thermodynamic basis to investigate transmetallation and solvent assisted linker exchange (SALE) methods and to synthesize new MOFs.
Co-reporter:Tae-Jin Park, Andrey A. Levchenko, Hongjun Zhou, Stanislaus S. Wong and Alexandra Navrotsky
Journal of Materials Chemistry A 2010 - vol. 20(Issue 39) pp:NaN8645-8645
Publication Date(Web):2010/09/08
DOI:10.1039/C0JM02192J
We report the direct determination of surface enthalpies for nanophase TiO2 anatase with different morphologies derived from drop solution calorimetry in a molten sodium molybdate (3Na2Oŀ4MoO3) solvent at 702 °C. The energetics of surface hydration has been measured using a Calvet microcalorimeter coupled with a gas dosing system. The surface enthalpies of hydrated surfaces for anatase TiO2 nanoparticles, nanowires and sea-urchin-like assemblies are 0.51 ± 0.05, 1.07 ± 0.28, and 1.29 ± 0.16 J m−2, respectively, whereas those of anhydrous surfaces are 0.74 ± 0.04, 1.24 ± 0.28, and 1.41 ± 0.16 J m−2, respectively. The trend in TiO2, which shows higher surface enthalpies for more complex nanostructures, is consistent with that reported in ZnO. The shape-dependent surface enthalpy at the nanoscale level is discussed in terms of exposed surface structures. The enthalpies of hydration appear to be similar for all morphologies.
Co-reporter:Gustavo Carneiro Cardoso da Costa, Lili Wu and Alexandra Navrotsky
Journal of Materials Chemistry A 2011 - vol. 21(Issue 6) pp:NaN1845-1845
Publication Date(Web):2010/12/08
DOI:10.1039/C0JM03090B
The energetics of the (1 − x)PMN–xPT solid solutions have been investigated using high temperature oxide melt solution calorimetry in 3Na2O·4MoO3solvent at 702 °C. The solid solutions show positive heats of mixing, reflecting the changes in structure from cubic to tetragonal to monoclinic and the morphotropic phase transition. The synthesis of a perovskite phase without a secondary pyrochlore phase depends on the annealing temperature and time. The enthalpies of reactions involved in synthesis and decomposition of the PMN perovskite were measured for the first time by oxide melt solution calorimetry. The decomposition of perovskite to pyrochlore plus MgO and PbO is energetically favorable, as is the formation of perovskite from columbite and lead oxide.
Co-reporter:A. V. Radha, J. D. Furman, M. Ati, B. C. Melot, J. M. Tarascon and A. Navrotsky
Journal of Materials Chemistry A 2012 - vol. 22(Issue 46) pp:NaN24452-24452
Publication Date(Web):2012/08/29
DOI:10.1039/C2JM34071B
Isothermal acid solution calorimetry was employed to investigate the relative thermochemical stabilities of two polymorphs of the LiFe1−xMnxSO4F (0 ≤ x ≤ 1) solid solution series: triplite and tavorite. These compounds have shown promise as lithium-ion battery cathodes, and a fuller understanding of their thermodynamics will aid in synthesis and their practical application. The linear energetic trends among triplites and tavorites indicate greater stabilization of each of these structures from the binary components with increase in manganese content and suggest a negligible heat of mixing of Fe and Mn ions. The tavorite phase, formed for x < 0.2, appears energetically more stable than the triplite. The formation of the disordered triplite structure appears to be entropy driven, and the factors that increase the disorder of the system (e.g. rapid phase formation) favor the triplite structure. Further, the free energy change associated with the tavorite to triplite transformation obtained by calculating configurational entropies using measured enthalpies was almost zero (−1.3 ± 0.8 kJ mol−1) at ambient temperature but becomes exothermic at 500 °C (−4.3 ± 0.8 to −6.8 ± 0.8 kJ mol−1). This suggests that both tavorite and triplite (with random cation distribution) are equally stable at ambient temperature but the tavorite to triplite transformation is thermodynamically favored at high temperatures because of entropy.
Co-reporter:M. D. Gonçalves, P. S. Maram, R. Muccillo and A. Navrotsky
Journal of Materials Chemistry A 2014 - vol. 2(Issue 42) pp:NaN17847-17847
Publication Date(Web):2014/09/05
DOI:10.1039/C4TA03487B
The enthalpies of formation from binary oxide components at 25 °C of Ba(Zr1−xYx)O3−δ, x = 0.1 to 0.5 solid solutions are measured by high temperature oxide melt solution calorimetry in a molten solvent, 3Na2O·4MoO3 at 702 °C. The enthalpy of formation is exothermic for all the compositions and becomes less negative when increasing yttrium content from undoped (−115.12 ± 3.69 kJ mol−1) to x = 0.5 (−77.09 ± 4.31 kJ mol−1). The endothermic contribution to the enthalpy of formation with doping content can be attributed to lattice distortions related to the large ionic radius difference of yttrium and zirconium and vacancy formation. For 0.3 ≤ x ≤ 0.5, the enthalpy of formation appears to level off, consistent with an exothermic contribution from defect clustering. Raman spectra indicate changes in short range structural features as a function of dopant content and, suggests that from x = 0.3 to 0.5 the defects begins to cluster significantly in the solid solution, which corroborates with the thermodynamic data and the drop-off in proton conductivity from x > 0.3.
Co-reporter:A. V. Radha, C. V. Subban, M. L. Sun, J. M. Tarascon and A. Navrotsky
Journal of Materials Chemistry A 2014 - vol. 2(Issue 19) pp:NaN6894-6894
Publication Date(Web):2014/03/27
DOI:10.1039/C3TA15457B
The thermodynamic stabilities of lithium hydroxysulfates of general formula LiMSO4OH (M = Co, Fe, Mn) with layered and tavorite structures have been investigated using isothermal acid solution calorimetry. These compounds have been explored as sustainable F-free alternatives to F-based flurosulfate cathode materials. The energetic trends for layered LiMSO4OH (M = Co, Fe and Mn) samples generally showed a decrease in stability with an increase in ionic radius (Co2+ to Mn2+), reflecting weaker M–O bonds and increasing structural distortions. The low symmetry tavorite LiFeSO4OH with a structure containing corner-shared octahedral chains is less stable than layered LiFeSO4OH with a more symmetric edge-shared octahedral structure. Structural distortions within the metal octahedra as well as changes in sulfate bonding and symmetry of the SO42− groups appear to control the thermodynamic and electrochemical behavior of LiMSO4OH (M = Co, Fe and Mn) materials. Both redox potential and thermodynamic stability of layered LiMSO4OH (M = Co, Fe and Mn) can be correlated to the lowering of the sulfate bonding symmetry in the structure from C3v to C2v.
Co-reporter:D. Wu, T. M. McDonald, Z. Quan, S. V. Ushakov, P. Zhang, J. R. Long and A. Navrotsky
Journal of Materials Chemistry A 2015 - vol. 3(Issue 8) pp:NaN4254-4254
Publication Date(Web):2015/01/14
DOI:10.1039/C4TA06496H
For coordinatively unsaturated metal–organic frameworks (MOFs), the metal centers can be functionalized as CO2 capture/storage adsorbents by grafting species having specific active groups. We report direct measurement of enthalpy of adsorption of CO2 on an alkylamine-appended MOF, mmen-Mg2(dobpdc) employing gas adsorption calorimetry at 298, 323 and 348 K. This methodology provides, for the first time, the detailed dependence of energy and entropy of sorption as a function of coverage and temperature. The enthalpy data suggest three types of adsorption events: strongest exothermic initial chemisorption at low coverage, majority moderate chemisorption at intermediate loading and weakest physisorption at highest coverage. The partial molar properties and isotherms are consistent with the presence of two different potential chemisorption mechanisms: 2:1 (amine–CO2) stoichiometry near zero coverage and 1:1 afterwards. Both chemical potential and differential enthalpy of adsorption become less negative with increasing temperature, implying increasing adsorbent entropy at elevated temperature. These observations are consistent with weaker CO2 binding at higher temperature.
Co-reporter:A. V. Radha, L. Lander, G. Rousse, J. M. Tarascon and A. Navrotsky
Journal of Materials Chemistry A 2015 - vol. 3(Issue 6) pp:NaN2608-2608
Publication Date(Web):2014/12/17
DOI:10.1039/C4TA05066E
Rational design and development of new Li-polyanion battery materials by exercising synthetic control have led to a new class of Li2M(SO4)2 compounds in monoclinic (M = Mn, Fe, Co) and orthorhombic (M = Fe, Co, Ni) polymorphic forms, using solid state (ceramic) and ball milling methods respectively. The enthalpies of formation from binary sulfates determined using isothermal acid solution calorimetry are positive, and show decrease in energetic metastability with increase in ionic radius for both monoclinic and orthorhombic (except for Ni) polymorphs. The higher symmetry orthorhombic polymorphs with Fe and Co are energetically less stable than the corresponding monoclinic polymorphs. The vibrational/rotational disorder of the SO4 tetrahedra is identified as the most likely cause of the entropy term (TΔS) of the free energy to overcome the positive enthalpy of formation in monoclinic and orthorhombic phases. Driven by thermodynamic metastability, the orthorhombic Li2M(SO4)2 (M = Fe, Co) polymorphs transform irreversibly into the monoclinic phase on heat treatment. Orthorhombic Li2Ni(SO4)2, formed by a ceramic route is thermodynamically stable and does not transform to the monoclinic phase on heating. The formation of metastable orthorhombic samples by ball milling is consistent with earlier thermodynamic studies on other Li-hydroxy/fluorosulfate systems, for which metastable tavorite polymorphs could be formed only by mild chemical synthetic approaches. This work demonstrates that the entropy term can play a key role for the synthesis, stability and phase transformation among polymorphs of Li-polyanionic compounds.
Co-reporter:Sulata Kumari Sahu, Speranta Tanasescu, Barbara Scherrer, Cornelia Marinescu and Alexandra Navrotsky
Journal of Materials Chemistry A 2015 - vol. 3(Issue 38) pp:NaN19496-19496
Publication Date(Web):2015/08/21
DOI:10.1039/C5TA03655K
Lanthanide cobalt perovskites LnCoO3−δ (Ln = La, Nd, Sm, and Gd) are important materials for electroceramics, catalysts, and electrodes in solid oxide fuel cells. Formation enthalpies of LnCoO3−δ compounds were measured using high temperature oxide melt solution calorimetry. The formation enthalpies of LaCoO2.992, NdCoO2.985, SmCoO2.982 and GdCoO2.968 from constituent binary oxides (Ln2O3, CoO) and O2 gas are −111.87 ± 1.36, −98.49 ± 1.33, −91.56 ± 1.46 and −88.16 ± 1.45 kJ mol−1, respectively. Thus these perovskites become energetically less stable with decrease in ionic radius of the lanthanide (from La to Gd), which corresponds to a decreasing tolerance factor and increasing oxygen deficiency. The thermodynamic stability of LaCoO2.992, NdCoO2.985, SmCoO2.982 and GdCoO2.968 was also assessed considering their oxygen partial pressures for decomposition, with good agreement between thermochemical and equilibrium data.
Co-reporter:X. Guo, Zs. Rak, A. H. Tavakoli, U. Becker, R. C. Ewing and A. Navrotsky
Journal of Materials Chemistry A 2014 - vol. 2(Issue 40) pp:NaN16954-16954
Publication Date(Web):2014/08/14
DOI:10.1039/C4TA03683B
The thermodynamic stability of Th-doped yttrium iron garnet (Y3Fe5O12, YIG) as a possible actinide-bearing material has been investigated using calorimetric measurements and first-principles electronic-structure calculations. Yttrium iron garnet with thorium substitution ranging from 0.04 to 0.07 atoms per formula unit (Y3−xThxFe5O12, x = 0.04–0.07) was synthesized using a citrate–nitrate combustion method. High-temperature oxide melt solution calorimetry was used to determine their enthalpy of formation. The thermodynamic analysis demonstrates that, although the substitution enthalpy is slightly endothermic, an entropic driving force for the substitution of Th for Y leads to a near-zero change in the Gibbs free energy. First-principles calculations within the density functional theory (DFT) indicate that the main limiting factors for Th incorporation into the YIG structure are the narrow stability domain of the host YIG and the formation of ThO2 as a secondary phase. Nevertheless, the defect formation energy calculations suggest that by carefully tuning the atomic and electronic chemical potentials, Th can be incorporated into YIG. The thermodynamic results, as a whole, support the possible use of garnet phases as nuclear waste forms; however, this will require careful consideration of the repository conditions.
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 6) pp:NaN2337-2337
Publication Date(Web):2013/12/11
DOI:10.1039/C3CP54553A
Rare-earth stabilized bismuth oxides are known for their excellent ionic conductivity at intermediate temperatures. However, previous studies have shown that their conductivity deteriorates during extended heat treatments at 500–600 °C, although the fluorite phase is maintained. In this study, the enthalpies of formation of quenched and aged ytterbia- and dysprosia-stabilized bismuth oxides were measured using high-temperature oxide melt solution calorimetry in 3Na2O–4MoO3 solvent at 702 °C. While a modest energy difference (−2 to −3 kJ mol−1) drives the kinetically slow aging transformation in the ytterbia-stabilized system at moderate dopant contents, no energetic driving force is detectable in the dysprosia-stabilized system. Although the small magnitude of the exothermic ordering energy suggests extensive short range ordering in both the quenched and aged samples, the anion configuration specific to the aged samples is nevertheless responsible for the significant decrease in conductivity.
.jpg)


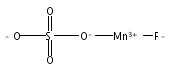
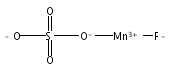





























![[1,1'-Biphenyl]-4,4'-dicarboxylic acid, 3,3'-dihydroxy-](/data/chemimg/102600/861533-46-2.png)
![[1,1'-Biphenyl]-4,4'-dicarboxylic acid, 3,3'-dihydroxy-](/data/chemimg/102600/861533-46-2_b.png)






![Benzene, 1,4-dibromo-2,5-bis[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]-](http://img.cochemist.com/ccimg/187800/187754-76-3.png)
![Benzene, 1,4-dibromo-2,5-bis[2-[2-(2-methoxyethoxy)ethoxy]ethoxy]-](http://img.cochemist.com/ccimg/187800/187754-76-3_b.png)
![Poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene]](http://img.cochemist.com/ccimg/138200/138184-36-8.png)
![Poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene]](http://img.cochemist.com/ccimg/138200/138184-36-8_b.png)
![Ethanol, 2,2'-[1,5-naphthalenediylbis(oxy-2,1-ethanediyloxy)]bis-](/data/chemimg/106300/136133-14-7.png)
![Ethanol, 2,2'-[1,5-naphthalenediylbis(oxy-2,1-ethanediyloxy)]bis-](/data/chemimg/106300/136133-14-7_b.png)





![Ferrate(4-), chloro[[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-κN21,κN22,κN23,κN24)tetrakis[benzoato]](6-)]-, hydrogen (1:4), (SP-5-12)-](/data/chemimg/99300/55266-17-6.png)
![Ferrate(4-), chloro[[4,4',4'',4'''-(21H,23H-porphine-5,10,15,20-tetrayl-κN21,κN22,κN23,κN24)tetrakis[benzoato]](6-)]-, hydrogen (1:4), (SP-5-12)-](/data/chemimg/99300/55266-17-6_b.png)












![Ethanol, 2,2'-[1,5-naphthalenediylbis(oxy-2,1-ethanediyloxy-2,1-ethanediyloxy-2,1-ethanediyloxy)]bis-, 1,1'-bis(4-methylbenzenesulfonate)](/data/chemimg/3776600/158394-33-3.png)
![Ethanol, 2,2'-[1,5-naphthalenediylbis(oxy-2,1-ethanediyloxy-2,1-ethanediyloxy-2,1-ethanediyloxy)]bis-, 1,1'-bis(4-methylbenzenesulfonate)](/data/chemimg/3776600/158394-33-3_b.png)



































![Copper, triaqua[μ-[1,3,5-benzenetricarboxylato(3-)-κO1:κO'1]][μ -[1,3,5-benzenetricarboxylato(3-)-κO1:κO3:κO'1]]tri-, compd. with N,N-](/data/chemimg/122200/222404-03-7.png)
![Copper, triaqua[μ-[1,3,5-benzenetricarboxylato(3-)-κO1:κO'1]][μ -[1,3,5-benzenetricarboxylato(3-)-κO1:κO3:κO'1]]tri-, compd. with N,N-](/data/chemimg/122200/222404-03-7_b.png)