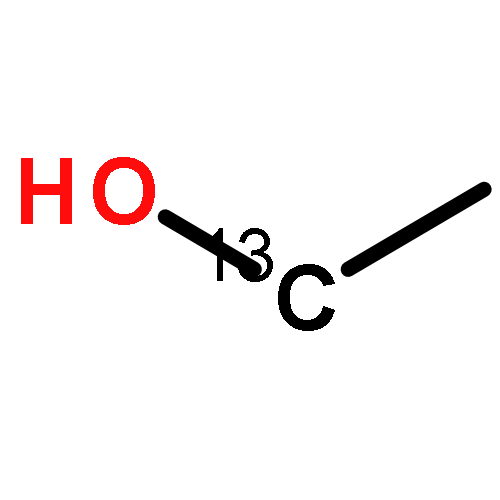
Co-reporter:Gang-hua Deng;Yuneng Shen;Zhigang He;Qiang Zhang;Bo Jiang;Guorong Wu;Xueming Yang
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 6) pp:4345-4351
Publication Date(Web):2017/02/08
DOI:10.1039/C6CP07380H
In this report, ultrafast time-resolved infrared spectroscopy is used to study the rotational motion of the liquid ethanol molecule. The results showed that the methyl, methylene, and CO groups have close rotational relaxation times, 1–2 ps, and the rotational relaxation time of the hydroxyl group (–OH) is 8.1 ps. The fast motion of the methyl, methylene and CO groups, and the slow motion of the hydroxyl group suggested that the ethanol molecules experience anisotropic motion in the liquid phase. The slow motion of the hydroxyl group also shows that the hydrogen bonded network could be considered as an effective molecule. The experimental data provided in this report are helpful for theorists to build models to understand the molecular rotational motion of liquid ethanol. Furthermore, our experimental method, which can provide more data concerning the rotational motion of sub groups of liquid molecules, will be useful for understanding the complicated molecular motion in the liquid phase.
Co-reporter:Yuneng Shen, Gang-Hua Deng, Chuanqi Ge, Yuhuan Tian, Guorong Wu, Xueming Yang, Junrong Zheng and Kaijun Yuan
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 22) pp:14867-14873
Publication Date(Web):11 May 2016
DOI:10.1039/C6CP02878K
Herein, we discuss the study of solvation dynamics of lithium–succinonitrile (SN) plastic crystalline electrolytes by ultrafast vibrational spectroscopy. The infrared absorption spectra indicated that the CN stretch of the Li+ bound and unbound succinonitrile molecules in a same solution have distinct vibrational frequencies (2276 cm−1vs. 2253 cm−1). The frequency difference allowed us to measure the rotation decay times of solvent molecules bound and unbound to Li+ ion. The Li+ coordination number of the Li+–SN complex was found to be 2 in the plastic crystal phase (22 °C) and 2.5–3 in the liquid phase (80 °C), which is independent of the concentration (from 0.05 mol kg−1 to 2 mol kg−1). The solvation structures along with DFT calculations of the Li+–SN complex have been discussed. In addition, the dissociation percentage of lithium salt was also determined. In 0.5 mol kg−1 LiBF4–SN solutions at 80 °C, 60% ± 10% of the salt dissociates into Li+, which is bound by 2 or 3 solvent molecules. In the 0.5 mol kg−1 LiClO4–SN solutions at 80 °C, the salt dissociation ratio can be up to 90% ± 10%.
Co-reporter:Shu Su, Yvonne Dorenkamp, Shengrui Yu, Alec M. Wodtke, Dongxu Dai, Kaijun Yuan and Xueming Yang
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 22) pp:15399-15405
Publication Date(Web):06 May 2016
DOI:10.1039/C6CP01956K
Photodissociation dynamics of HBr at a series of photolysis wavelengths in the range of 123.90–125.90 nm and at around 137.0 nm have been studied using the H atom Rydberg “tagging” time-of-flight technique. The branching fractions between the channels forming ground Br(2P3/2) and spin–orbit excited Br(2P1/2) atoms together with the angular distributions of the products corresponding to these two channels have been measured. The photolysis wavelengths in this work excited the HBr molecule from the ground state X 1Σ+ to various Rydberg states and the V 1Σ+ ion-pair valence state. Predissociation via these states displays rich behavior, indicating the influence of the nature of initially excited states and the coupling to other bound or repulsive states on the predissociation dynamics.
Co-reporter:Chuanqi Ge, Yuneng Shen, Gang-Hua Deng, Yuhuan Tian, Dongqi Yu, Xueming Yang, Kaijun Yuan, and Junrong Zheng
The Journal of Physical Chemistry B 2016 Volume 120(Issue 12) pp:3187-3195
Publication Date(Web):March 11, 2016
DOI:10.1021/acs.jpcb.5b12652
Isotopic effects on the formation and dissociation kinetics of hydrogen bonds are studied in real time with ultrafast chemical exchange spectroscopy. The dissociation time of hydrogen bond between phenol-OH and p-xylene (or mesitylene) is found to be identical to that between phenol-OD and p-xylene (or mesitylene) in the same solvents. The experimental results demonstrate that the isotope substitution (D for H) has negligible effects on the hydrogen bond kinetics. DFT calculations show that the isotope substitution does not significantly change the frequencies of vibrational modes that may be along the hydrogen bond formation and dissociation coordinate. The zero point energy differences of these modes between hydrogen bonds with OH and OD are too small to affect the activation energy of the hydrogen bond dissociation in a detectible way at room temperature.
Co-reporter:Shengrui Yu, Shu Su, Dongxu Dai, Kaijun Yuan and Xueming Yang
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 15) pp:9659-9665
Publication Date(Web):14 Aug 2014
DOI:10.1039/C4CP02734E
The state-to-state dynamics of high-n Rydberg H-atom scattering with para-H2 at the collision energies of 0.45 and 1.07 eV have been studied using the H-atom Rydberg tagging time-of-flight technique. Both the inelastic scattering and reactive scattering are observed in the experimental time-of-flight spectra. The products H2(v′, j′ = odd) come only from reactive scattering and present clearly forward–backward asymmetric angular distributions, which differ from those of the corresponding ion–molecule reaction. The products H2(v′, j′ = even), however, come from both reactive scattering and inelastic scattering. Simulating the rotational distribution from reactive scattering, we found that most of the H2(v′, j′ = even) products come from inelastic scattering. The angular distributions of the product H2(v′, j′ = even) are consistent with what is predicted by the conventional textbook mechanism of inelastic scattering, and are a little different from those of the corresponding ion–molecule inelastic scattering. These results suggest that the effect of Rydberg electron could not be neglected in describing the differential cross sections of H* + para-H2 scattering. From the simulation, the branching ratios of the inelastic scattering channel were determined to be 66% and 79% at the collision energies of 0.45 and 1.07 eV, respectively.
Co-reporter:Yuneng Shen, Tianmin Wu, Bo Jiang, Ganghua Deng, Jiebo Li, Hailong Chen, Xunmin Guo, Chuanqi Ge, Yajing Chen, Jieya Hong, Xueming Yang, Kaijun Yuan, Wei Zhuang, and Junrong Zheng
The Journal of Physical Chemistry B 2015 Volume 119(Issue 30) pp:9893-9904
Publication Date(Web):July 2, 2015
DOI:10.1021/acs.jpcb.5b04530
In this work, MD simulations with two different force fields, vibrational energy relaxation and resonant energy transfer experiments, and neutron scattering data are used to investigate ion pairing and clustering in a series of GdmSCN aqueous solutions. The MD simulations reproduce the major features of neutron scattering experimental data very well. Although no information about ion pairing or clustering can be obtained from the neutron scattering data, MD calculations clearly demonstrate that substantial amounts of ion pairs and small ion clusters (subnanometers to a few nanometers) do exist in the solutions of concentrations 0.5 M*, 3 M*, and 5 M* (M* denotes mole of GdmSCN per 55.55 mole of water). Vibrational relaxation experiments suggest that significant amounts of ion pairs form in the solutions. Experiments measuring the resonant energy transfers among the thiocyanate anions in the solutions suggest that the ions form clusters and in the clusters the average anion distance is 5.6 Å (5.4 Å) in the 3 M* (5 M*) Gdm–DSCN/D2O solution.
Co-reporter:Hongzhen Wang, Shengrui Yu, Shu Su, Dongxu Dai, Kaijun Yuan, and Xueming Yang
The Journal of Physical Chemistry A 2015 Volume 119(Issue 46) pp:11313-11319
Publication Date(Web):October 22, 2015
DOI:10.1021/acs.jpca.5b08865
The state-selective photodissociation of diacetylene (C4H2) was studied in the wavelength range of 127.5–164.4 nm by high-resolution Rydberg H atom time-of-flight spectroscopy measurements. In the wavelength region, two Rydberg series nR and nR′ were state-selectively excited using tunable vacuum-ultraviolet laser radiation. In all photolysis wavelengths, two decay channels with different dissociation dynamics were observed. In one channel, the characteristic and isotropic translational energy distributions with a peak around 1800 cm–1 can be found, suggesting statistical dissociation through internal conversion (IC) from the Rydberg state to the ground state and then dissociation on the ground-state surface. In contrast to this, in the second channel, nonstatistical and anisotropic translational energy distributions were observed, possibly through IC to the excited repulsive state. The vibrational progressions of C4H (A2Π) products have also been observed and assigned to the CCC bend and C≡C stretch progressions in the second channel at 3R Rydberg states.
Co-reporter:Shengrui Yu, Shu Su, Yvonne Dorenkamp, Alec M. Wodtke, Dongxu Dai, Kaijun Yuan, and Xueming Yang
The Journal of Physical Chemistry A 2013 Volume 117(Issue 46) pp:11673-11678
Publication Date(Web):March 15, 2013
DOI:10.1021/jp312793k
Photodissociation dynamics of HNCO at photolysis wavelengths between 200 and 240 nm have been studied using the H-atom Rydberg tagging time-of-flight technique. Product translational energy distributions and angular distributions have been determined. At low photon energy excitation, the product translational energy distribution is nearly statistical and the angular distribution is isotropic, which is consistent with an indirect dissociation mechanism, i.e., internal conversion from S1 to S0 surface and dissociation on S0 surface. As the photon energy increases, a direct dissociation pathway on S1 surface opens up. The product translational energy distribution appears to be quite nonstatistical and the product angular distribution is anisotropic. The fraction of direct dissociation pathway is determined to be 36 ± 5% at 202.67 nm photolysis. Vibrational structures are observed in both direct and indirect dissociation pathways, which can be assigned to the NCO bending mode excitation with some stretching excitation.
Co-reporter:Shengrui Yu, Shu Su, Dongxu Dai, Kaijun Yuan, and Xueming Yang
The Journal of Physical Chemistry A 2013 Volume 117(Issue 50) pp:13564-13571
Publication Date(Web):September 16, 2013
DOI:10.1021/jp407556k
Photodissociation dynamics of the H-atom channel from HNCO photolysis between 124 and 137 nm have been studied using the H-atom Rydberg tagging time-of-flight technique. Product translational energy distributions and angular distributions have been determined. Two dissociation channels, H + NCO (X2Π) and H + NCO(A2Σ+), have been observed. The former channel involves two different dissociation pathways; one is a slow predissociation pathway through internal conversion from the excited state to the S0 state, and the other is a fast predissociation pathway through internal conversion from the excited state to the S1 state. The latter channel dominates by a prompt dissociation via coupling to the S2 state. As the photon energy increases, dissociation on the ground state S0 becomes dominant. Vibrational structures are observed in both the NCO(X) and NCO(A) channels, which can be assigned to the bending mode excitation with some stretching vibrational excitation.
Co-reporter:Shengrui Yu, Kaijun Yuan, Hui Song, Xin Xu, Dongxu Dai, Dong H. Zhang and Xueming Yang
Chemical Science 2012 vol. 3(Issue 9) pp:2839-2842
Publication Date(Web):25 Jun 2012
DOI:10.1039/C2SC20489D
Full quantum-state resolved differential cross-sections of the H*(n) + o-D2 → HD + D*(n′) reaction have been measured for the first time using the Rydberg H-atom time-of-flight method. Experimental results show that the angular distributions of HD product rotational states show a strong preference for forward scattering. This result is considerably different to that predicted by full quantum mechanical calculations on the corresponding ion–molecule reaction, suggesting that the ionic core and Rydberg electron coupling cannot be neglected in the Rydberg H-atom reactive scattering with D2 and, therefore, that the Fermi independent-collider model is not valid in describing the dynamics of Rydberg atom reactions with molecules.
Co-reporter:Shengrui Yu, Shu Su, Kaijun Yuan, Dongxu Dai, and Xueming Yang
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 17) pp:2420-2424
Publication Date(Web):August 15, 2012
DOI:10.1021/jz3010255
The state-resolved differential cross sections for the Rydberg-atom (RA) inelastic scattering process H*(n = 46) + O2(v = 0, j = 1,3) → H*(n′) + O2(v′, j′) have been measured by using the H-atom Rydberg tagging time-of-flight (HRTOF) technique. Extensive vibrational excitation of O2 products has been observed at the two collision energies of 0.64 and 1.55 eV. Experimental results show that the O2 products in the low vibrationally excited states are clearly forward-scattered, whereas those in the highly vibrationally excited states are mainly backward-scattered. Partially resolved rotational structures were also observed and assigned. The striking observation of extremely high energy transfer from translational to vibrational excitation at the backward direction could be explained involving charge transfer between proton and O2 molecule and possibly complex formation during the scattering process.Keywords: differential cross sections; inelastic scattering; Rydberg atom;
Co-reporter:Lina Cheng, Kaijun Yuan, Yuan Cheng, Qing Guo, Tao Wang, Dongxu Dai, and Xueming Yang and Richard N. Dixon
The Journal of Physical Chemistry A 2011 Volume 115(Issue 9) pp:1500-1507
Publication Date(Web):January 19, 2011
DOI:10.1021/jp109169f
The dissociation dynamics of HOD via two-photon excitation to the C̃ state have been investigated using the H-atom Rydberg tagging time-of-flight (TOF) technique. The H-atom action spectrum for the C̃ ← X̃ transition shows resolved rotational structure. Product translational energy distributions and angular distributions have also been recorded for the H + OD channel for three excited levels each with ka′ = 2. From these distributions, quantum state distributions and angular anisotropy parameters (β2 and β4) for the OD product were determined. These results are consistent with the nonadiabatic predissociation picture illustrated in the one-photon dissociation process for H2O. The heterogeneous dissociation pathway via Coriolis coupling is the dominant dissociation process in the present study. A high proportion of the total available energy is deposited into the rotational energy of the OD product. The anisotropic recoil distributions reveal the distinctive contributions from the alignment of the excited states and the dissociation process. Comparisons are also made between the results for HOD and H2O via the equivalent rotational transitions. The OH bond energy, Do(H−OD), of the HOD molecule is also determined to be 41283.0 ± 5 cm−1.
Co-reporter:Yongwei Zhang, Kaijun Yuan, Shengrui Yu and Xueming Yang
The Journal of Physical Chemistry Letters 2010 Volume 1(Issue 2) pp:475-479
Publication Date(Web):December 22, 2009
DOI:10.1021/jz900303e
Photodissociation of CH4 has been studied using the high-resolution Rydberg tagging time-of-flight technique. The TOF spectra show an important single C−H bond fission channel with partially resolved sharp features. Careful simulations indicate that these sharp peaks are due to highly rotationally excited CH3 products, which are likely produced through a conical intersection dissociation pathway between the excited and ground potential energy surfaces. The energy-dependent anisotropy parameter of the CH3 product has also been determined at various photolysis wavelengths. The results of this work show that the conical intersection between the S1 and S0 surfaces plays an essential role in the photochemistry of CH4.Keywords (keywords): conical intersection; methane photochemistry; nonadiabatic dissociation;
Co-reporter:Yuneng Shen, Gang-Hua Deng, Chuanqi Ge, Yuhuan Tian, Guorong Wu, Xueming Yang, Junrong Zheng and Kaijun Yuan
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 22) pp:NaN14873-14873
Publication Date(Web):2016/05/11
DOI:10.1039/C6CP02878K
Herein, we discuss the study of solvation dynamics of lithium–succinonitrile (SN) plastic crystalline electrolytes by ultrafast vibrational spectroscopy. The infrared absorption spectra indicated that the CN stretch of the Li+ bound and unbound succinonitrile molecules in a same solution have distinct vibrational frequencies (2276 cm−1vs. 2253 cm−1). The frequency difference allowed us to measure the rotation decay times of solvent molecules bound and unbound to Li+ ion. The Li+ coordination number of the Li+–SN complex was found to be 2 in the plastic crystal phase (22 °C) and 2.5–3 in the liquid phase (80 °C), which is independent of the concentration (from 0.05 mol kg−1 to 2 mol kg−1). The solvation structures along with DFT calculations of the Li+–SN complex have been discussed. In addition, the dissociation percentage of lithium salt was also determined. In 0.5 mol kg−1 LiBF4–SN solutions at 80 °C, 60% ± 10% of the salt dissociates into Li+, which is bound by 2 or 3 solvent molecules. In the 0.5 mol kg−1 LiClO4–SN solutions at 80 °C, the salt dissociation ratio can be up to 90% ± 10%.
Co-reporter:Shu Su, Yvonne Dorenkamp, Shengrui Yu, Alec M. Wodtke, Dongxu Dai, Kaijun Yuan and Xueming Yang
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 22) pp:NaN15405-15405
Publication Date(Web):2016/05/06
DOI:10.1039/C6CP01956K
Photodissociation dynamics of HBr at a series of photolysis wavelengths in the range of 123.90–125.90 nm and at around 137.0 nm have been studied using the H atom Rydberg “tagging” time-of-flight technique. The branching fractions between the channels forming ground Br(2P3/2) and spin–orbit excited Br(2P1/2) atoms together with the angular distributions of the products corresponding to these two channels have been measured. The photolysis wavelengths in this work excited the HBr molecule from the ground state X 1Σ+ to various Rydberg states and the V 1Σ+ ion-pair valence state. Predissociation via these states displays rich behavior, indicating the influence of the nature of initially excited states and the coupling to other bound or repulsive states on the predissociation dynamics.
Co-reporter:Shengrui Yu, Shu Su, Dongxu Dai, Kaijun Yuan and Xueming Yang
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 15) pp:NaN9665-9665
Publication Date(Web):2014/08/14
DOI:10.1039/C4CP02734E
The state-to-state dynamics of high-n Rydberg H-atom scattering with para-H2 at the collision energies of 0.45 and 1.07 eV have been studied using the H-atom Rydberg tagging time-of-flight technique. Both the inelastic scattering and reactive scattering are observed in the experimental time-of-flight spectra. The products H2(v′, j′ = odd) come only from reactive scattering and present clearly forward–backward asymmetric angular distributions, which differ from those of the corresponding ion–molecule reaction. The products H2(v′, j′ = even), however, come from both reactive scattering and inelastic scattering. Simulating the rotational distribution from reactive scattering, we found that most of the H2(v′, j′ = even) products come from inelastic scattering. The angular distributions of the product H2(v′, j′ = even) are consistent with what is predicted by the conventional textbook mechanism of inelastic scattering, and are a little different from those of the corresponding ion–molecule inelastic scattering. These results suggest that the effect of Rydberg electron could not be neglected in describing the differential cross sections of H* + para-H2 scattering. From the simulation, the branching ratios of the inelastic scattering channel were determined to be 66% and 79% at the collision energies of 0.45 and 1.07 eV, respectively.
Co-reporter:Gang-hua Deng, Yuneng Shen, Zhigang He, Qiang Zhang, Bo Jiang, Kaijun Yuan, Guorong Wu and Xueming Yang
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 6) pp:NaN4351-4351
Publication Date(Web):2017/01/18
DOI:10.1039/C6CP07380H
In this report, ultrafast time-resolved infrared spectroscopy is used to study the rotational motion of the liquid ethanol molecule. The results showed that the methyl, methylene, and CO groups have close rotational relaxation times, 1–2 ps, and the rotational relaxation time of the hydroxyl group (–OH) is 8.1 ps. The fast motion of the methyl, methylene and CO groups, and the slow motion of the hydroxyl group suggested that the ethanol molecules experience anisotropic motion in the liquid phase. The slow motion of the hydroxyl group also shows that the hydrogen bonded network could be considered as an effective molecule. The experimental data provided in this report are helpful for theorists to build models to understand the molecular rotational motion of liquid ethanol. Furthermore, our experimental method, which can provide more data concerning the rotational motion of sub groups of liquid molecules, will be useful for understanding the complicated molecular motion in the liquid phase.
Co-reporter:Shengrui Yu, Kaijun Yuan, Hui Song, Xin Xu, Dongxu Dai, Dong H. Zhang and Xueming Yang
Chemical Science (2010-Present) 2012 - vol. 3(Issue 9) pp:NaN2842-2842
Publication Date(Web):2012/06/25
DOI:10.1039/C2SC20489D
Full quantum-state resolved differential cross-sections of the H*(n) + o-D2 → HD + D*(n′) reaction have been measured for the first time using the Rydberg H-atom time-of-flight method. Experimental results show that the angular distributions of HD product rotational states show a strong preference for forward scattering. This result is considerably different to that predicted by full quantum mechanical calculations on the corresponding ion–molecule reaction, suggesting that the ionic core and Rydberg electron coupling cannot be neglected in the Rydberg H-atom reactive scattering with D2 and, therefore, that the Fermi independent-collider model is not valid in describing the dynamics of Rydberg atom reactions with molecules.