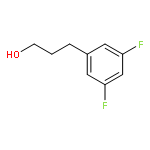
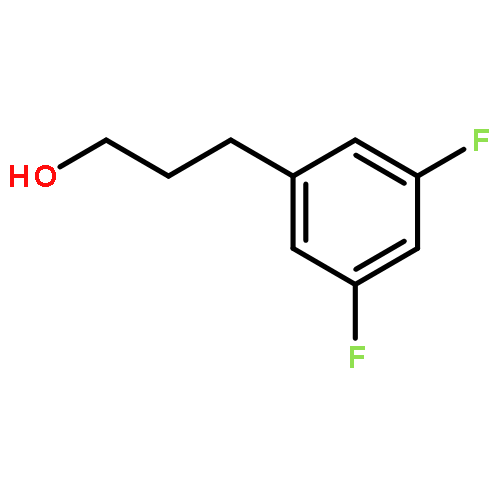
Co-reporter:Atsushi Kato ; Erina Hayashi ; Saori Miyauchi ; Isao Adachi ; Tatsushi Imahori ; Yoshihiro Natori ; Yuichi Yoshimura ; Robert J. Nash ; Hideyuki Shimaoka ; Izumi Nakagome ; Jun Koseki ; Shuichi Hirono
Journal of Medicinal Chemistry 2012 Volume 55(Issue 23) pp:10347-10362
Publication Date(Web):October 29, 2012
DOI:10.1021/jm301304e
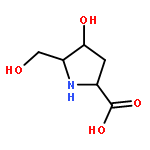
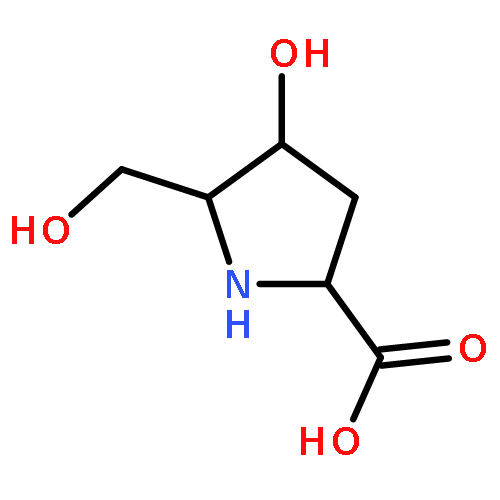
We report on the synthesis and the biological evaluation of a series of α-1-C-alkylated 1,4-dideoxy-1,4-imino-l-arabinitol (LAB) derivatives. The asymmetric synthesis of the derivatives was achieved by asymmetric allylic alkylation, ring-closing metathesis, and Negishi cross-coupling as key reactions. α-1-C-Butyl-LAB is a potent inhibitor of intestinal maltase, isomaltase, and sucrase, with IC50 values of 0.13, 4.7, and 0.032 μM, respectively. Matrix-assisted laser desorption ionization time-of-flight mass spectrometric analysis revealed that this compound differs from miglitol in that it does not influence oligosaccharide processing and the maturation of glycoproteins. A molecular docking study of maltase-glucoamylase suggested that the interaction modes and the orientations of α-1-C-butyl-LAB and miglitol are clearly different. Furthermore, α-1-C-butyl-LAB strongly suppressed postprandial hyperglycemia at an early phase, similar to miglitol in vivo. It is noteworthy that the effective dose was about 10-fold lower than that for miglitol. α-1-C-Butyl-LAB therefore represents a new class of promising compounds that can improve postprandial hyperglycemia.
Co-reporter:Yoshihiro Natori, Tatsushi Imahori, Keiichi Murakami, Yuichi Yoshimura, Shinpei Nakagawa, Atsushi Kato, Isao Adachi, Hiroki Takahata
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 2) pp:738-741
Publication Date(Web):15 January 2011
DOI:10.1016/j.bmcl.2010.11.112
The asymmetric synthesis of 1-C-alkyl-l-arabinoiminofuranoses 1 was achieved by asymmetric allylic alkylation (AAA), ring closing metathesis (RCM), and Negishi cross coupling as key reactions. Some of the prepared compounds showed potent inhibitory activities towards intestinal maltase, with IC50 values comparable to those of commercial drugs such as acarbose, voglibose, and miglitol, which are used in the treatment of type 2 diabetes. Among them, the inhibitory activity (IC50 = 0.032 μM) towards intestinal sucrase of 1c was quite strong compared to the above commercial drugs.
Co-reporter:Yukako Saito, Yuichi Yoshimura, Hiroki Takahata
Tetrahedron Letters 2010 Volume 51(Issue 52) pp:6915-6917
Publication Date(Web):29 December 2010
DOI:10.1016/j.tetlet.2010.10.114
The chemoselective O-tert-butoxycarbonylation of phenols using low levels (5–0.1 mol %) of 6,7-dimethoxyisoquinoline as a reusable organocatalyst is described.
Co-reporter:Yuichi Yoshimura, Chiaki Ohara, Tatsushi Imahori, Yukako Saito, Atsushi Kato, Saori Miyauchi, Isao Adachi, Hiroki Takahata
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 17) pp:8273-8286
Publication Date(Web):1 September 2008
DOI:10.1016/j.bmc.2008.06.016
We have synthesized 3-hydroxy- and 3,4,5-trihydroxypipecolic acid derivatives corresponding to 5-aza derivatives of uronic acids and evaluated their inhibitory activities against various glycosidases including β-glucuronidase. Compounds 4 and 5 were chosen as common intermediates for the synthesis of 3,4,5-trihydroxypipecolic acids and 3-hydroxypipecolic acids as well as for 3-hydroxybaikiain, a unique natural product isolated from a toxic mushroom. Cross aldol reaction of N-Boc-allylglycine derivative with acrolein followed by the ring-closing metathesis gave 4 and 5 as a mixture of diastereomers which could be separated by silica gel column chromatography. By employing lipase-catalyzed kinetic resolution, the synthesis of both l- and d-isomers of 3,4,5-trihydroxy- and 3-hydroxypipecolic acids was achieved. None of the compounds tested showed inhibitory activity against α- and β-glucosidases. On the other hand, l-23 and l-29 were found to have potent inhibitory activity against β-glucuronidase. In addition, it is interesting that some uronic-type azasugar derivatives showed moderate inhibitory activities against β-N-acetylglucosaminidase.
Co-reporter:Chiaki Ohara, Ryouko Takahashi, Tatsunori Miyagawa, Yuichi Yoshimura, Atsushi Kato, Isao Adachi, Hiroki Takahata
Bioorganic & Medicinal Chemistry Letters 2008 Volume 18(Issue 6) pp:1810-1813
Publication Date(Web):15 March 2008
DOI:10.1016/j.bmcl.2008.02.028
A highly practicable synthesis of both enantiomers of 3-hydroxypipecolic acid derivatives 1, 2, 3, 4 is described. Screening of these molecules for glycosidase inhibition has been examined. Compound 3 was shown to be a potent inhibitor of β-N-acetylglucosaminidase as well as Escherichia coli β-glucuronidase.
Co-reporter:Hiroki Takahata;Yumiko Suto;Erina Kato;Yuichi Yoshimura;Hidekazu Ouchi
Advanced Synthesis & Catalysis 2007 Volume 349(Issue 4-5) pp:
Publication Date(Web):20 MAR 2007
DOI:10.1002/adsc.200600559
The palladium-catalyzed deracemization of N-protected alkyl carbonates of 5-hydroxy-3-piperidenes by use of chiral phosphine ligands is described. A Trost ligand such as (R)-BPA was found to be a suitable chiral ligand for the deracemization, providing N-protected 5-hydroxy-3-piperidenes in good yields with good to high enantioselectivities. A plausible mechanism for the reaction is proposed.
Co-reporter:Hiroki Takahata, Yukako Saito and Motohiro Ichinose
Organic & Biomolecular Chemistry 2006 vol. 4(Issue 8) pp:1587-1595
Publication Date(Web):14 Mar 2006
DOI:10.1039/B601489E
A novel C2-symmetric 2,6-diallylpiperidine carboxylic acid methyl ester 1 was prepared by the double asymmetric allylboration of glutaldehyde followed by an aminocyclization and carbamation. On the basis of desymmetrization of 1 using iodocarbamation, one allyl group of 1 was protected and monofunctionalizations of the resulting oxazolidinone 11 were performed. The reaction of the N-methoxycarbonyl piperidine 25 employing decarbamation reagent (n-PrSLi or TMSI) as a key step gave oxazolidinone 26 or 17 including an intramolecular ring formation, which was transformed in a few steps into (−)-porantheridine (2) and (−)-2-epi-porantheridine (3), respectively. In addition, the expedient synthesis of (+)-epi-dihydropinidine (4), (2R,6R)-trans-solenopsin A (5), and precoccinelline (6), starting from 11 is described.



![Benzene, 1,1'-[(2Z)-2-butene-1,4-diylbis(oxymethylene)]bis[4-methoxy-](http://img.cochemist.com/ccimg/179600/179548-99-3.png)
![Benzene, 1,1'-[(2Z)-2-butene-1,4-diylbis(oxymethylene)]bis[4-methoxy-](http://img.cochemist.com/ccimg/179600/179548-99-3_b.png)