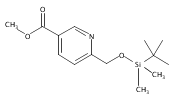
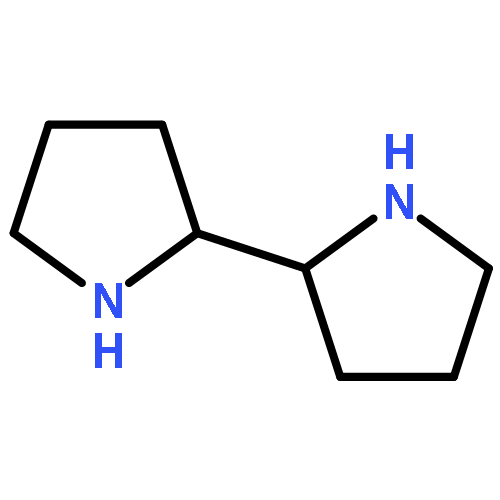
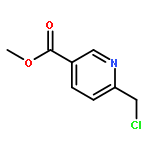
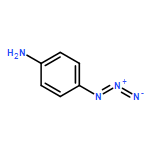
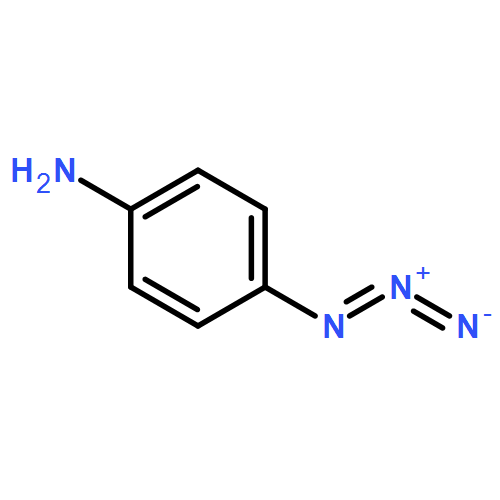
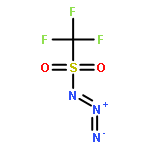
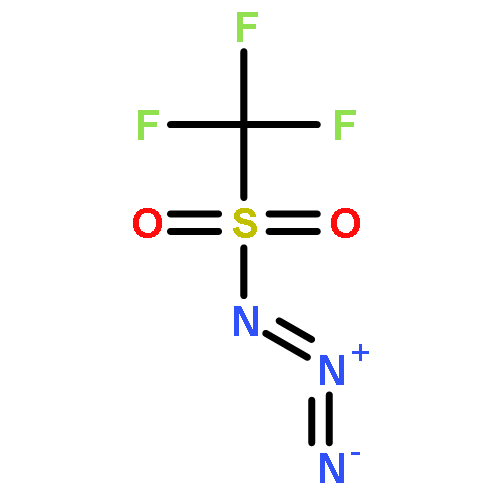
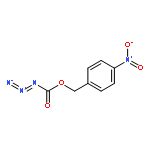
Co-reporter:Cristina Rosés, Cristina Camó, Kristy Vogels, Marta Planas, and Lidia Feliu
Organic Letters 2016 Volume 18(Issue 16) pp:4140-4143
Publication Date(Web):August 5, 2016
DOI:10.1021/acs.orglett.6b02281
The first solid-phase strategy for the synthesis of cyclic depsipeptides containing a phenyl ester linkage in their structure is described. The key steps of the synthesis were the formation of the phenyl ester bond and the on-resin head-to-side-chain cyclization. The amino acid configuration significantly influenced the formation and the stability of the cyclic depsipeptides. The presence of a l-Tyr1 and a d-Tyr7 led to the most stable sequences.
Co-reporter:Marta Soler, Marta González-Bártulos, Eduard Figueras, Xavi Ribas, Miquel Costas, Anna Massaguer, Marta Planas and Lidia Feliu
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 5) pp:1470-1480
Publication Date(Web):26 Nov 2014
DOI:10.1039/C4OB01875C
The undecapeptide KKLFKKILKKL-NH2 (BP16) is a non-toxic cell-penetrating peptide (CPP) that is mainly internalized into cancer cells through a clathrin dependent endocytic mechanism and localizes in late endosomes. Moreover, this CPP is able to enhance the cellular uptake of chlorambucil (CLB) improving its cytotoxicity. In this work, we further explored the cell-penetrating properties of BP16 and those of its arginine analogue BP308. We investigated the influence on the cytotoxicity and on the cellular uptake of conjugating CLB at the N- or the C-terminal end of these undecapeptides. The effect of incorporating the cathepsin B-cleavable sequence Gly-Phe-Leu-Gly in CLB-BP16 and CLB-BP308 conjugates was also evaluated. The activity of CLB was significantly improved when conjugated at the N- or the C-terminus of BP16, or at the N-terminus of BP308. While CLB alone was not active (IC50 of 73.7 to >100 μM), the resulting conjugates displayed cytotoxic activity against CAPAN-1, MCF-7, PC-3, 1BR3G and SKMEL-28 cell lines with IC50 values ranging from 8.7 to 25.5 μM. These results were consistent with the internalization properties observed for the corresponding 5(6)-carboxyfluorescein-labeled conjugates. The presence of the tetrapeptide Gly-Phe-Leu-Gly at either the N- or the C-terminus of CLB-BP16 conjugates further increased the efficacy of CLB (IC50 of 3.6 to 16.2 μM), which could be attributed to its selective release in the lysosomal compartment. Enzymatic assays with cathepsin B showed the release of CLB-Gly-OH from these sequences within a short time. Therefore, the combination of BP16 with an enzymatic cleavable sequence can be used as a drug delivery system for the effective uptake and release of drugs in cancer cells.
Co-reporter:Sílvia Vilà;Esther Badosa;Emilio Montesinos;Lidia Feliu
European Journal of Organic Chemistry 2015 Volume 2015( Issue 5) pp:1117-1129
Publication Date(Web):
DOI:10.1002/ejoc.201403344
Abstract
The solid-phase conjugation of the antimicrobial peptide c(KKLKKFKKLQ) (BPC194) to a linear or cyclic sequence through a 1,2,3-triazole ring is described. Cyclic alkynyl-peptidyl resins derived from BPC194 were treated with azidopeptides derived from the antimicrobial peptide BP100 or from the bacteriocin iturin A. The cyclic alkynyl-peptidyl resins incorporated at the 3-position a propargylglycine, a glutamic acid residue derivatized with propargylamine or a lysine bearing a propioloyl group. The reactions of the cyclic alkynyl resins with the BP100-derived azidopeptides depended on the length and the sequence of the azidopeptides. The reactions were performed by treatment of the alkynyl resin with CuI and ascorbic acid, and required the presence of piperidine/DMF or DIEA in 2,6-lutidine/DMF. The latter conditions also allowed the conjugation of the alkynyl-peptidyl resin bearing a propioloyl lysine to a linear or cyclic azidopeptide derived from the cyclic moiety of iturin A.
Co-reporter:Marta Soler, Marta González-Bártulos, David Soriano-Castell, Xavi Ribas, Miquel Costas, Francesc Tebar, Anna Massaguer, Lidia Feliu and Marta Planas
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 10) pp:1652-1663
Publication Date(Web):10 Jan 2014
DOI:10.1039/C3OB42422G
Antimicrobial peptides are an interesting source of non-cytotoxic drug delivery vectors. Herein, we report on the identification of a new cell-penetrating peptide (KKLFKKILKKL-NH2, BP16) from a set of antimicrobial peptides selected from a library of cecropin-melittin hybrids (CECMEL11) previously designed to be used in plant protection. This set of peptides was screened for their cytotoxicity against breast adenocarcinoma MCF-7, pancreas adenocarcinoma CAPAN-1 and mouse embryonic fibroblast 3T3 cell lines. BP16 resulted to be non-toxic against both malignant and non-malignant cells at concentrations up to 200 μM. We demonstrated by flow cytometry and confocal microscopy that BP16 is mainly internalized in the cells through a clathrin dependent endocytosis and that it efficiently accumulates in the cell cytoplasm. We confirmed that the cell-penetrating properties of BP16 are retained after conjugating it to the breast tumor homing peptide CREKA. Furthermore, we assessed the potential of BP16 as a drug delivery vector by conjugating the anticancer drug chlorambucil to BP16 and to a CREKA-BP16 conjugate. The efficacy of the drug increased between 6 and 9 times when conjugated to BP16 and between 2 and 4.5 times when attached to the CREKA-BP16 derivative. The low toxicity and the excellent cell-penetrating properties clearly suggest that BP16 is a suitable vector for the delivery of therapeutic agents into cells.
Co-reporter:Sílvia Vilà;Cristina Camó;Eduard Figueras;Esther Badosa;Emilio Montesinos;Lidia Feliu
European Journal of Organic Chemistry 2014 Volume 2014( Issue 22) pp:4785-4794
Publication Date(Web):
DOI:10.1002/ejoc.201402111
Abstract
The solid-phase synthesis of cyclic lipopeptidotriazoles derived from the cyclic decapeptide c(Lys-Lys2-Leu-Lys-Lys5-Phe-Lys-Lys-Leu-Gln) (BPC194), incorporating a triazolyl ring at Lys2 and a hexanoyl group at Lys5, was studied. Four different strategies that required the use of five orthogonal protecting groups (Fmoc, tBu, All, pNZ, ivDde) were explored. The influence of the side-chain protection of Lys2 and Lys5 with the ivDde and pNZ groups was evaluated by incorporating Lys2(ivDde)/Lys5(pNZ) or Lys2(pNZ)/Lys5(ivDde). The order of removal of these protecting groups and of the introduction of the hexanoyl and triazolyl moieties was also studied. The best strategy included: (i) synthesis of a cyclic peptidyl resin bearing Lys2(ivDde) and Lys5(pNZ); (ii) pNZ group removal; (iii) acylation with hexanoic acid; (iv) ivDde group removal; and (v) acylation with propiolic acid followed by an azide–alkyne 1,3-dipolar cycloaddition. By using this protocol, a set of cyclic lipopeptidotriazoles was prepared in high purities.
Co-reporter:Sílvia Vilà, Esther Badosa, Emilio Montesinos, Lidia Feliu and Marta Planas
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 20) pp:3365-3374
Publication Date(Web):20 Mar 2013
DOI:10.1039/C3OB40319J
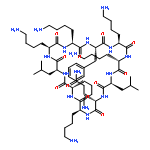
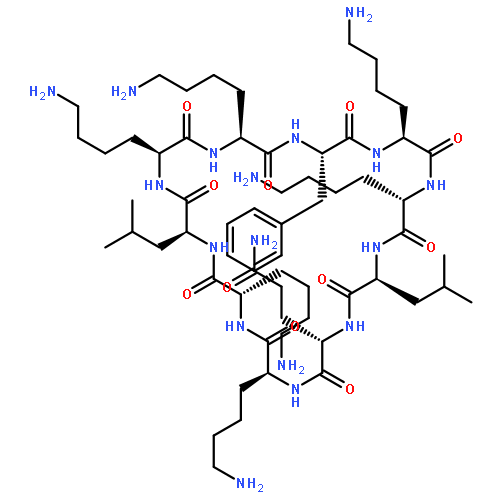
A concise solid-phase synthesis of cyclic lipopeptides derived from the antimicrobial peptide c(Lys-Lys-Leu-Lys-Lys-Phe-Lys-Lys-Leu-Gln) (BPC194) was accomplished. Three different synthetic routes were explored. Best results were obtained using a protocol that includes as key steps: (i) synthesis of the cyclic peptidyl resin incorporating the Lys residue to be acylated protected at the Nε-amino group with an ivDde group, (ii) selective removal of the ivDde group, and (iii) acylation. These compounds were screened for their in vitro growth inhibition of bacterial and fungal phytopathogens and for their cytotoxic effects on eukaryotic cells. A sequence with high antimicrobial activity and low hemolysis was identified, constituting a good candidate for the design of new antimicrobial agents.
Co-reporter:Imma Güell;Sílvia Vilà;Lluís Micaló;Esther Badosa;Emilio Montesinos;Lidia Feliu
European Journal of Organic Chemistry 2013 Volume 2013( Issue 22) pp:4933-4943
Publication Date(Web):
DOI:10.1002/ejoc.201300215
Abstract
Cyclic peptidotriazoles derived from the antimicrobial cyclic peptide c(Lys-Lys-Leu-Lys-Lys-Phe-Lys-Lys-Leu-Gln) (BPC194) were prepared by incorporating a triazolyl amino acid at the 3-position. The synthesis was accomplished on solid-phase and involved as the key step a copper-catalyzed cycloaddition reaction between a resin-bound alkyne or a resin-bound azide and a range of azides or alkynes in solution, respectively. This methodology was also applied to the synthesis of a conjugated peptide containing a cyclic and a linear peptidyl sequence linked through a triazolyl ring. Cyclic peptidotriazoles were obtained in excellent purities starting either from an alkynyl or an azido peptidyl resin. These compounds were screened in vitro for their growth inhibition of bacterial phytopathogens and for their cytotoxic effects on eukaryotic cells. Peptide sequences with high antibacterial activity and low hemolysis were identified, constituting good candidates for the design of new antimicrobial agents.
Co-reporter:Iteng Ng-Choi;Marta Soler;Vanessa Cerezo;Esther Badosa;Emilio Montesinos;Lidia Feliu
European Journal of Organic Chemistry 2012 Volume 2012( Issue 23) pp:4321-4332
Publication Date(Web):
DOI:10.1002/ejoc.201200291
Abstract
A microwave-assisted solid-phase Suzuki–Miyaura reaction has been employed for the synthesis of 5-arylhistidine-containing peptides. In particular, sequences containing a 5-arylhistidine at the 1- or 4-positions have been designed based on lead antimicrobial peptides. The cross-coupling involved the arylation of a resin-bound 5-bromohistidine with an arylboronic acid in solution under microwave irradiation. This protocol is compatible with common protecting groups used in peptide chemistry. The resulting biaryl linear undecapeptides were screened for their antibacterial, antifungal and hemolytic activities. The results showed that the presence of an imidazole ring significantly decreases the cytotoxicity.
Co-reporter:Ana Afonso;Olaf Cussó;Lidia Feliu
European Journal of Organic Chemistry 2012 Volume 2012( Issue 31) pp:6204-6211
Publication Date(Web):
DOI:10.1002/ejoc.201200832
Abstract
A concise and efficient solid-phase synthesis of biaryl cyclic peptides containing a Phe–Tyr or a Tyr–Tyr linkage has been accomplished. The key steps include a Miyaura borylation of a resin-bound 3-iodotyrosine and a microwave-assisted Suzuki–Miyaura reaction for the formation of the macrocycle. First, the feasibility of the solid-phase Miyaura borylation of a 3-iodotyrosyltripeptide was established. Then, the Suzuki–Miyaura reaction was applied to the cross-coupling of linear 3-boronotyrosine-containing peptidyl resins with iodobenzene and with halogenated aromatic amino acids. Finally, this methodology was extended to the synthesis of biaryl cyclic peptides through the preparation of a linear peptidyl resin containing both the required boronate and halogenated derivatives, followed by a microwave-assisted Suzuki–Miyaura macrocyclization.
Co-reporter:Ana Afonso, Lidia Feliu, Marta Planas
Tetrahedron 2011 67(12) pp: 2238-2245
Publication Date(Web):
DOI:10.1016/j.tet.2011.01.084
Co-reporter:Lidia Feliu, Glòria Oliveras, Anna D. Cirac, Emili Besalú, Cristina Rosés, Ramon Colomer, Eduard Bardají, Marta Planas, Teresa Puig
Peptides (November 2010) Volume 31(Issue 11) pp:2017-2026
Publication Date(Web):1 November 2010
DOI:10.1016/j.peptides.2010.07.027
Antimicrobial peptides have been considered as potential candidates for cancer therapy. We report here the cytotoxicity of a library of 66 antibacterial cyclodecapeptides on human carcinoma cell lines, and their effects on apoptosis [as assessed by cleavage of poly(ADP-ribose) polymerase (PARP)] and cell signaling proteins (p53 and ERK1/2) in cultured human cervical carcinoma cells. A design of experiments approach permitted to analyze the results of a subset of 16 peptides and define rules for high anticancer activity against MDA-MB-231 breast carcinoma cells. Eight peptides were identified with IC50 values ranging from 18.5 to 57.5 μM against the five cell lines tested, being HeLa cells the most sensitive. Among these sequences, BPC88, BPC96, BPC98, and BPC194 displayed specificity and high cytotoxicity against HeLa cells (IC50 of 22.5–38.5 μM), showed low hemolytic activity and low cytotoxicity to non-malignant fibroblasts, and were stable to proteases in human serum. Induction of apoptosis by these peptides was observed and the apoptotic effect of BPC88 and BPC96 caused a marked decrease on the activated form of ERK1/2 kinase and an induction of p53. We further showed that BPC96 at low doses synergized the cytotoxic effect of cisplatin. These findings suggest that cyclic decapeptides may represent novel anticancer agents providing a new strategy in cancer therapy.Research highlights▶ Anticancer activity of a library of cyclic antimicrobial decapeptides. ▶ Induction of apoptosis by cyclic decapeptides. ▶ Synergism with cisplatin.
Co-reporter:Imma Güell, Lluís Micaló, Laura Cano, Esther Badosa, Rafael Ferre, Emilio Montesinos, Eduard Bardají, Lidia Feliu, Marta Planas
Peptides (January 2012) Volume 33(Issue 1) pp:9-17
Publication Date(Web):1 January 2012
DOI:10.1016/j.peptides.2011.12.003
We designed and prepared peptidotriazoles based on the antimicrobial peptide BP100 (LysLysLeuPheLysLysIleLeuLysTyrLeu-NH2) by introducing a triazole ring in the peptide backbone or onto the side chain of a selected residue. These compounds were screened for their in vitro growth inhibition of bacterial and fungal phytopathogens, and for their cytotoxic effects on eukaryotic cells and tobacco leaves. Their proteolytic susceptibility was also analyzed. The antibacterial activity and the hemolysis were influenced by the amino acid that was modified with the triazole as well as by the absence of presence of a substituent in this heterocyclic ring. We identified sequences active against the bacteria Xanthomonas axonopodis pv. vesicatoria, Erwinia amylovora, Pseudomonas syringae pv. syringae (MIC of 1.6–12.5 μM), and against the fungi Fusarium oxysporum (MIC < 6.2–12.5 μM) with low hemolytic activity (0–23% at 50 μM), high stability to protease digestion and no phytotoxicity. These peptidotriazoles constitute good candidates to design new antimicrobial agents.Graphical abstractDownload full-size imageHighlights► Peptidotriazoles with an optimal biological profile were identified. ► They showed high antimicrobial activity and stability, low hemolysis, and no phytotoxicity. ► The synthesis involved the formation of the 1,2,3-triazole by a click reaction.
Co-reporter:Sílvia Vilà, Esther Badosa, Emilio Montesinos, Lidia Feliu and Marta Planas
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 20) pp:NaN3374-3374
Publication Date(Web):2013/03/20
DOI:10.1039/C3OB40319J
A concise solid-phase synthesis of cyclic lipopeptides derived from the antimicrobial peptide c(Lys-Lys-Leu-Lys-Lys-Phe-Lys-Lys-Leu-Gln) (BPC194) was accomplished. Three different synthetic routes were explored. Best results were obtained using a protocol that includes as key steps: (i) synthesis of the cyclic peptidyl resin incorporating the Lys residue to be acylated protected at the Nε-amino group with an ivDde group, (ii) selective removal of the ivDde group, and (iii) acylation. These compounds were screened for their in vitro growth inhibition of bacterial and fungal phytopathogens and for their cytotoxic effects on eukaryotic cells. A sequence with high antimicrobial activity and low hemolysis was identified, constituting a good candidate for the design of new antimicrobial agents.
Co-reporter:Marta Soler, Marta González-Bártulos, Eduard Figueras, Xavi Ribas, Miquel Costas, Anna Massaguer, Marta Planas and Lidia Feliu
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 5) pp:NaN1480-1480
Publication Date(Web):2014/11/26
DOI:10.1039/C4OB01875C
The undecapeptide KKLFKKILKKL-NH2 (BP16) is a non-toxic cell-penetrating peptide (CPP) that is mainly internalized into cancer cells through a clathrin dependent endocytic mechanism and localizes in late endosomes. Moreover, this CPP is able to enhance the cellular uptake of chlorambucil (CLB) improving its cytotoxicity. In this work, we further explored the cell-penetrating properties of BP16 and those of its arginine analogue BP308. We investigated the influence on the cytotoxicity and on the cellular uptake of conjugating CLB at the N- or the C-terminal end of these undecapeptides. The effect of incorporating the cathepsin B-cleavable sequence Gly-Phe-Leu-Gly in CLB-BP16 and CLB-BP308 conjugates was also evaluated. The activity of CLB was significantly improved when conjugated at the N- or the C-terminus of BP16, or at the N-terminus of BP308. While CLB alone was not active (IC50 of 73.7 to >100 μM), the resulting conjugates displayed cytotoxic activity against CAPAN-1, MCF-7, PC-3, 1BR3G and SKMEL-28 cell lines with IC50 values ranging from 8.7 to 25.5 μM. These results were consistent with the internalization properties observed for the corresponding 5(6)-carboxyfluorescein-labeled conjugates. The presence of the tetrapeptide Gly-Phe-Leu-Gly at either the N- or the C-terminus of CLB-BP16 conjugates further increased the efficacy of CLB (IC50 of 3.6 to 16.2 μM), which could be attributed to its selective release in the lysosomal compartment. Enzymatic assays with cathepsin B showed the release of CLB-Gly-OH from these sequences within a short time. Therefore, the combination of BP16 with an enzymatic cleavable sequence can be used as a drug delivery system for the effective uptake and release of drugs in cancer cells.
Co-reporter:Marta Soler, Marta González-Bártulos, David Soriano-Castell, Xavi Ribas, Miquel Costas, Francesc Tebar, Anna Massaguer, Lidia Feliu and Marta Planas
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 10) pp:NaN1663-1663
Publication Date(Web):2014/01/10
DOI:10.1039/C3OB42422G
Antimicrobial peptides are an interesting source of non-cytotoxic drug delivery vectors. Herein, we report on the identification of a new cell-penetrating peptide (KKLFKKILKKL-NH2, BP16) from a set of antimicrobial peptides selected from a library of cecropin-melittin hybrids (CECMEL11) previously designed to be used in plant protection. This set of peptides was screened for their cytotoxicity against breast adenocarcinoma MCF-7, pancreas adenocarcinoma CAPAN-1 and mouse embryonic fibroblast 3T3 cell lines. BP16 resulted to be non-toxic against both malignant and non-malignant cells at concentrations up to 200 μM. We demonstrated by flow cytometry and confocal microscopy that BP16 is mainly internalized in the cells through a clathrin dependent endocytosis and that it efficiently accumulates in the cell cytoplasm. We confirmed that the cell-penetrating properties of BP16 are retained after conjugating it to the breast tumor homing peptide CREKA. Furthermore, we assessed the potential of BP16 as a drug delivery vector by conjugating the anticancer drug chlorambucil to BP16 and to a CREKA-BP16 conjugate. The efficacy of the drug increased between 6 and 9 times when conjugated to BP16 and between 2 and 4.5 times when attached to the CREKA-BP16 derivative. The low toxicity and the excellent cell-penetrating properties clearly suggest that BP16 is a suitable vector for the delivery of therapeutic agents into cells.

![L-Norleucine, 6-azido-N-[(9H-fluoren-9-ylmethoxy)carbonyl]-](/data/chemimg/116900/159610-89-6.png)
![L-Norleucine, 6-azido-N-[(9H-fluoren-9-ylmethoxy)carbonyl]-](/data/chemimg/116900/159610-89-6_b.png)