Co-reporter:Tian Shi, Hao Li, Sergei Tretiak, and Vladimir Y. Chernyak
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 22) pp:3946-3952
Publication Date(Web):October 28, 2014
DOI:10.1021/jz501912d
Effects of disorder and exciton–phonon interactions are the major factors controlling photoinduced dynamics and energy-transfer processes in conjugated organic semiconductors, thus defining their electronic functionality. All-atom quantum-chemical simulations are potentially capable of describing such phenomena in complex “soft” organic structures, yet they are frequently computationally restrictive. Here we efficiently characterize how electronic excitations in branched conjugated molecules interact with molecular distortions using the exciton scattering (ES) approach as a fundamental principle combined with effective tight-binding models. Molecule geometry deformations are incorporated to the ES view of electronic excitations by identifying the dependence of the Frenkel-type exciton Hamiltonian parameters on the characteristic geometry parameters. We illustrate our methodology using two examples of intermolecular distortions, bond length alternation and single bond rotation, which constitute vibrational degrees of freedom strongly coupled to the electronic system in a variety of conjugated systems. The effect on excited-state electronic structures has been attributed to localized variation of exciton on-site energies and couplings. As a result, modifications of the entire electronic spectra due to geometric distortions can be efficiently and accurately accounted for with negligible numerical cost. The presented approach can be potentially extended to model electronic structures and photoinduced processes in bulk amorphous polymer materials.Keywords: conjugated molecule; excited state; exciton scattering; exciton−phonon Hamiltonian; geometry distortion; tight binding model;
Co-reporter:Hao Li, Michael J. Catanzaro, Sergei Tretiak, and Vladimir Y. Chernyak
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 4) pp:641-647
Publication Date(Web):January 28, 2014
DOI:10.1021/jz4027198
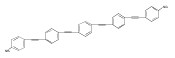
Attachment of chemical substituents (such as polar moieties) constitutes an efficient and convenient way to modify physical and chemical properties of conjugated polymers and oligomers. Associated modifications in the molecular electronic states can be comprehensively described by examining scattering of excitons in the polymer’s backbone at the scattering center representing the chemical substituent. Here, we implement effective tight-binding models as a tool to examine the analytical properties of the exciton scattering matrices in semi-infinite polymer chains with substitutions. We demonstrate that chemical interactions between the substitution and attached polymer are adequately described by the analytical properties of the scattering matrices. In particular, resonant and bound electronic excitations are expressed via the positions of zeros and poles of the scattering amplitude, analytically continued to complex values of exciton quasi-momenta. We exemplify the formulated concepts by analyzing excited states in conjugated phenylacetylenes substituted by perylene.Keywords: bound state; conjugation; electronic excitation; ES approach; perylene; resonant state; tight-binding model;
Co-reporter:Hao Li, Vladimir Y. Chernyak, and Sergei Tretiak
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 24) pp:3734-3739
Publication Date(Web):November 28, 2012
DOI:10.1021/jz301521p
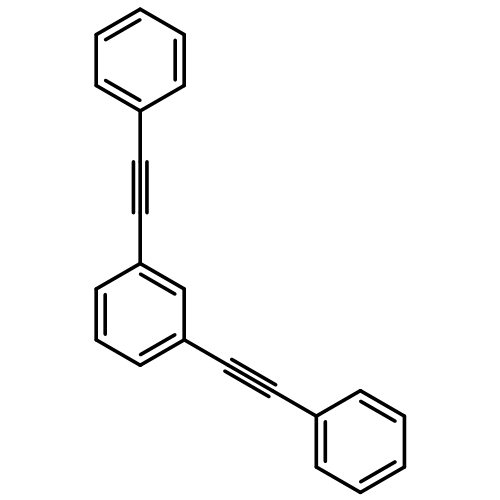
The exciton scattering (ES) method allows efficient calculations of spectroscopic observables in large low-dimensional conjugated molecular systems. To compute the transition dipoles between the ground and excited electronic states, we should extract the ES dipole parameters from quantum chemistry calculations in simple molecular fragments. In this manuscript, we show how to retrieve these parameters from any reference quantum chemistry model that uses an arbitrary nonorthogonal and possibly overcomplete atomic orbital basis set. Our approach relies on the natural atomic orbital (NAO) representation, in which the basis functions are orthonormal and the atom-like character is preserved. We apply the ES approach, combined with the NAO analysis to optical spectra of branched phenylacetylene oligomers. Absorption spectra predicted by the ES method demonstrate close agreement with the results of direct quantum chemistry calculations, when the Time-Dependent Density Functional Theory (TD-DFT) being used as a reference. This testifies applicability of a variety of quantum-chemical techniques, where the NAO population analysis can be conducted, for the ES framework.Keywords: branched structure; conjugated molecule; conjugation; electronic excitation; ES approach; transition charge; transition dipole moment;
Co-reporter:Hao Li, Chao Wu, Sergey V. Malinin, Sergei Tretiak, and Vladimir Y. Chernyak
The Journal of Physical Chemistry B 2011 Volume 115(Issue 18) pp:5465-5475
Publication Date(Web):December 31, 2010
DOI:10.1021/jp110317d
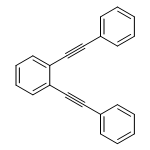
The capability of the exciton scattering approach, an efficient methodology for excited states in branched conjugated molecules, is extended to include symmetric triple and quadruple joints that connect linear segments on the basis of the phenylacetylene backbone. The obtained scattering matrices that characterize these vertices are used in application of our approach to several test structures, where we find excellent agreement with the transition energies computed by the reference quantum chemistry. We introduce topological charges, associated with the scattering matrices, which help to formulate useful relations between the number of excitations in the exciton band and the number of repeat units. The obtained features of the scattering phases are analyzed in terms of the observed excited state electronic structure.
Co-reporter:Hao Li, Chao Wu, Sergey V. Malinin, Sergei Tretiak, and Vladimir Y. Chernyak
The Journal of Physical Chemistry Letters 2010 Volume 1(Issue 23) pp:3396-3400
Publication Date(Web):November 16, 2010
DOI:10.1021/jz1013533
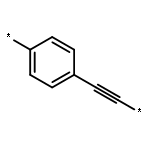
Quantum-chemical computations allow one to predict a variety of interesting electronic properties of donor and acceptor substituted conjugated molecules. However, the complexity of such systems often limits physical interpretation of the computations and makes them impossible in larger molecules. In this study, the exciton scattering (ES) methodology is extended to analyze the excited-state structure of donor and acceptor substituted conjugated oligomers. The extracted reflection phases, transition charge, and dipole parameters of the modified termini are used to quantify the influence of the substitution on the molecular electronic and optical spectra. In particular, intuitive relationships between the substituent’s electron withdrawing or donating ability and the ES parameters are discussed. A good agreement of the absorption spectra between the ES approach and the reference quantum-chemical computations demonstrates that the ES approach is qualified for such conjugated push−pull systems.Keywords (keywords): conjugation; dendrimers; electronic excitations; exciton reflection; substitution; transition density; transition dipoles;
Co-reporter:Andrei Piryatinski, Sergei Tretiak, Vladimir Y. Chernyak
Chemical Physics 2008 Volume 347(1–3) pp:25-38
Publication Date(Web):23 May 2008
DOI:10.1016/j.chemphys.2008.01.010
Abstract
Studying non-adiabatic effects in molecular dynamics simulations and modeling their optical signatures in linear and non-linear spectroscopies calls for electronic structure calculations in a situation when the ground state is degenerate or almost degenerate. Such degeneracy causes serious problems in invoking single Slater determinant Hartree–Fock (HF) and density functional theory (DFT) methods. To resolve this problem, we develop a generalization of time-dependent (dynamical) variational approach which accounts for the degenerate or almost degenerate ground state structure. Specifically, we propose a ground state ansatz for the subspace of generalized electronic configurations spanned on the degenerate grounds state multi-electron wavefunctions. Further employing the invariant form of Hamilton dynamics we arrive with the classical equations of motion describing the time-evolution of this subspace in the vicinity of the stationary point. The developed approach can be used for accurate calculations of molecular excited states and electronic spectra in the degenerate case.
Co-reporter:Chao Wu, Sergei Tretiak, Vladimir Y. Chernyak
Chemical Physics Letters 2007 Volume 433(4–6) pp:305-311
Publication Date(Web):12 January 2007
DOI:10.1016/j.cplett.2006.11.069
Optical properties of polar push–pull chromophore (diphenylpolyene with donor/acceptor terminal substituents) are studied using hybrid time-dependent density functional theory (TD-DFT). The optical transitions are thoroughly examined. This includes one- and two-photon absorption, and fluorescence, as a function of the underlying density functional, the basis set choice, and the solvent. Calculated excited state properties are found to be strongly dependent on the density functional model used. Hybrid approximations with small fractions of the orbital exchange (e.g. B3LYP) strongly favor zwitterionic-type states with pronounced charge-transfer character. Models with large percentage of orbital exchange (e.g. BHandHLYP) result in neutral-base electronic excitations.Calculated spectra of polar push–pull chromophore (diphenylpolyene with donor/acceptor terminal substituents) using hybrid time-dependent density functional theory. One- and two-photon absorption, and fluorescence are studied as a function of the density functional, the basis set choice, and the solvent.
Co-reporter:Hao Li, Chao Wu, Sergey V. Malinin, Sergei Tretiak, Vladimir Y. Chernyak
Chemical Physics (20 December 2016) Volume 481() pp:
Publication Date(Web):20 December 2016
DOI:10.1016/j.chemphys.2016.08.033
The exciton scattering (ES) technique is a multiscale approach based on the concept of a particle in a box and developed for efficient calculations of excited-state electronic structure and optical spectra in low-dimensional conjugated macromolecules. Within the ES method, electronic excitations in molecular structure are attributed to standing waves representing quantum quasi-particles (excitons), which reside on the graph whose edges and nodes stand for the molecular linear segments and vertices, respectively. Exciton propagation on the linear segments is characterized by the exciton dispersion, whereas exciton scattering at the branching centers is determined by the energy-dependent scattering matrices. Using these ES energetic parameters, the excitation energies are then found by solving a set of generalized “particle in a box” problems on the graph that represents the molecule. Similarly, unique energy-dependent ES dipolar parameters permit calculations of the corresponding oscillator strengths, thus, completing optical spectra modeling. Both the energetic and dipolar parameters can be extracted from quantum-chemical computations in small molecular fragments and tabulated in the ES library for further applications. Subsequently, spectroscopic modeling for any macrostructure within a considered molecular family could be performed with negligible numerical effort. We demonstrate the ES method application to molecular families of branched conjugated phenylacetylenes and ladder poly-para-phenylenes, as well as structures with electron donor and acceptor chemical substituents. Time-dependent density functional theory (TD-DFT) is used as a reference model for electronic structure. The ES calculations accurately reproduce the optical spectra compared to the reference quantum chemistry results, and make possible to predict spectra of complex macromolecules, where conventional electronic structure calculations are unfeasible.
Co-reporter:Jérémy R. Rouxel, Vladimir Y. Chernyak and Shaul Mukamel
Chemical Science (2010-Present) 2016 - vol. 7(Issue 11) pp:NaN6831-6831
Publication Date(Web):2016/07/11
DOI:10.1039/C6SC01743F
A spatially non-local response tensor description of linear chiral signals such as circular dichroism is developed. By working directly with the vector potential rather that the electric and magnetic fields, we recast the signals in terms of correlation functions of charge and current densities and avoid the tedious expansion in multipoles.