Co-reporter:Nicholas D. Eastham, Alexander S. Dudnik, Thomas J. Aldrich, Eric F. Manley, Thomas J. Fauvell, Patrick E. Hartnett, Michael R. Wasielewski, Lin X. Chen, Ferdinand S. Melkonyan, Antonio Facchetti, Robert P. H. Chang, and Tobin J. Marks
Chemistry of Materials May 23, 2017 Volume 29(Issue 10) pp:4432-4432
Publication Date(Web):May 3, 2017
DOI:10.1021/acs.chemmater.7b00964
Perylenediimide (PDI) small molecule acceptor (SMA) crystallinity and donor polymer aggregation and crystallinity effects on bulk-heterojunction microstructure and polymer solar cell (PSC) performance are systematically investigated. Two high-performance polymers, semicrystalline poly[5-(2-hexyldodecyl)-4H-thieno[3,4-c]pyrrole-4,6(5H)-dione-1,3-yl-alt-4,4″dodecyl-2,2′:5′,2″-terthiophene-5,5″-diyl] (PTPD3T or D1) and amorphous poly{4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl-alt-(4-(2-ethylhexyl)-3-fluorothieno[3,4-b]thiophene-2-carboxylate-2,6-diyl) (PBDTT-FTTE or D2), are paired with three PDI-based SMAs (A1–A3) of differing crystallinity (A1 is the most, A3 is the least crystalline). The resulting PSC performance trends are strikingly different from those of typical fullerene-based PSCs and are highly material-dependent. The present trends reflect synergistic aggregation propensities between the SMA and polymer components. Importantly, the active layer morphology is templated by the PDI in some blends and by the polymer in others, with the latter largely governed by the polymer aggregation. Thus, PTPD3T templating capacity increases as self-aggregation increases (greater Mn), optimizing PSC performance with A2, while A3-based cells exhibit an inverse relationship between polymer aggregation and performance, which is dramatically different from fullerene-based PSCs. For PBDTT-FTTE, A2-based cells again deliver the highest PCEs of ∼5%, but here both A2 and PBDTT-FTTE (medium Mn) template the morphology. Overall, the present results underscore the importance of nonfullerene acceptor aggregation for optimizing PSC performance and offer guidelines for pairing SMAs with acceptable donor polymers.
Co-reporter:Thomas J. Fauvell, Tianyue Zheng, Nicholas E. Jackson, Mark A. Ratner, Luping Yu, and Lin X. Chen
Chemistry of Materials 2016 Volume 28(Issue 8) pp:2814
Publication Date(Web):April 8, 2016
DOI:10.1021/acs.chemmater.6b00734
Organic semiconductors have garnered substantial interest in optoelectronics, but their device performances exhibit strong dependencies on material crystallinity and packing. In an effort to understand the interactions dictating the morphological and photophysical properties of a high-performing photovoltaic polymer, PTB7, a series of short oligomers and low molecular weight polymers of PTB7 were synthesized. Chain-length dependent optical studies of these oligomers demonstrate that PTB7’s low-energy visible absorption is largely due to self-aggregation-induced ordering, rather than in-chain charge transfer, as previously thought. By examining molecular weight and concentration dependent optical properties, supplemented by molecular dynamics simulations, we attribute polymeric PTB7’s unique midgap fluorescence and concentration independent absorption spectrum to an interplay between low molecular weight unaggregated strands and high-molecular weight self-aggregated (folded) strands. Specifically, we propose that the onset of PTB7 self-folding occurs between 7 and 13 repeat units, but the aggregates characteristic of polymeric PTB7 only develop at lengths of ∼30 repeat units. Atomistic molecular dynamics simulations of PTB7 corroborate these conclusions, and a simple relation is proposed which quantifies the free-energy of conjugated polymer folding. This study provides detailed guidance in the design of intra- and interchain contributions to the photophysical and morphological properties of polymeric semiconductors.
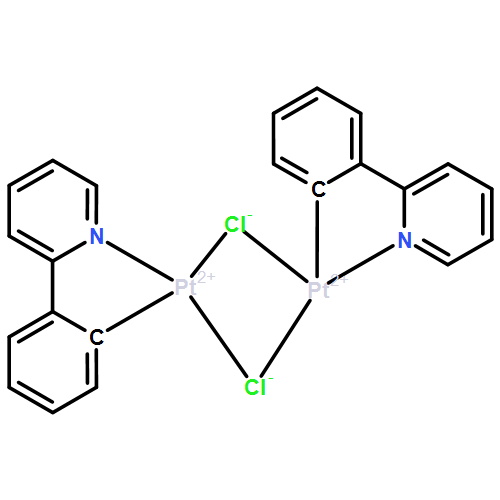
Co-reporter:Samantha E. Brown-Xu, Matthew S. J. Kelley, Kelly A. Fransted, Arnab Chakraborty, George C. Schatz, Felix N. Castellano, and Lin X. Chen
The Journal of Physical Chemistry A 2016 Volume 120(Issue 4) pp:543-550
Publication Date(Web):January 13, 2016
DOI:10.1021/acs.jpca.5b11233
The influence of molecular structure on excited-state properties and dynamics of a series of cyclometalated platinum dimers was investigated through a combined experimental and theoretical approach using femtosecond transient absorption (fs TA) spectroscopy and density functional theory (DFT) calculations. The molecules have the general formula [Pt(ppy)(μ-R2pz)]2, where ppy = 2-phenylpyridine, pz = pyrazolate, and R = H, Me, Ph, or tBu, and are strongly photoluminescent at room temperature. The distance between the platinum centers in this A-frame geometry can be varied depending on the steric bulk of the bridging pyrazolate ligands that exert structural constraints and compress the Pt–Pt distance. At large Pt–Pt distances there is little interaction between the subunits, and the chromophore behaves similar to a monomer with excited states described as mixtures of ligand-centered and metal-to-ligand charge transfer (LC/MLCT) transitions. When the Pt(II) centers are brought closer together with bulky bridging ligands, they interact through their dz2 orbitals and the S1 and T1 states are best characterized as metal–metal-to-ligand charge transfer (MMLCT) in character. The results of the femtoseconds TA experiments reveal that intersystem crossing (ISC) occurs on ultrafast time scales (τS1 < 200 fs), while there are two relaxation processes occurring within the triplet manifold, τ1 = 0.5–3.2 ps and τ2 = 20–70 ps; the longer time constants correspond to the presence of bulkier bridging ligands. DFT calculations illustrate that the Pt–Pt distances further contract in the T1 3MMLCT states; therefore, slower relaxation may be related to a larger structural reorganization. Subsequent investigations using faster time resolution are planned to measure the ISC process as well as to identify any potential coherent interaction(s) between the platinum centers that may occur.
Co-reporter:Nanjia Zhou; Alexander S. Dudnik; Ting I. N. G. Li; Eric F. Manley; Thomas J. Aldrich; Peijun Guo; Hsueh-Chung Liao; Zhihua Chen; Lin X. Chen; Robert P. H. Chang; Antonio Facchetti; Monica Olvera de la Cruz;Tobin J. Marks
Journal of the American Chemical Society 2015 Volume 138(Issue 4) pp:1240-1251
Publication Date(Web):December 31, 2015
DOI:10.1021/jacs.5b10735
The influence of the number-average molecular weight (Mn) on the blend film morphology and photovoltaic performance of all-polymer solar cells (APSCs) fabricated with the donor polymer poly[5-(2-hexyldodecyl)-1,3-thieno[3,4-c]pyrrole-4,6-dione-alt-5,5-(2,5-bis(3-dodecylthiophen-2-yl)thiophene)] (PTPD3T) and acceptor polymer poly{[N,N′-bis(2-octyldodecyl)naphthalene-1,4,5,8-bis(dicarboximide)-2,6-diyl]-alt-5,5′-(2,2′-bithiophene)} (P(NDI2OD-T2); N2200) is systematically investigated. The Mn effect analysis of both PTPD3T and N2200 is enabled by implementing a polymerization strategy which produces conjugated polymers with tunable Mns. Experimental and coarse-grain modeling results reveal that systematic Mn variation greatly influences both intrachain and interchain interactions and ultimately the degree of phase separation and morphology evolution. Specifically, increasing Mn for both polymers shrinks blend film domain sizes and enhances donor–acceptor polymer–polymer interfacial areas, affording increased short-circuit current densities (Jsc). However, the greater disorder and intermixed feature proliferation accompanying increasing Mn promotes charge carrier recombination, reducing cell fill factors (FF). The optimized photoactive layers exhibit well-balanced exciton dissociation and charge transport characteristics, ultimately providing solar cells with a 2-fold PCE enhancement versus devices with nonoptimal Mns. Overall, it is shown that proper and precise tuning of both donor and acceptor polymer Mns is critical for optimizing APSC performance. In contrast to reports where maximum power conversion efficiencies (PCEs) are achieved for the highest Mns, the present two-dimensional Mn optimization matrix strategy locates a PCE “sweet spot” at intermediate Mns of both donor and acceptor polymers. This study provides synthetic methodologies to predictably access conjugated polymers with desired Mn and highlights the importance of optimizing Mn for both polymer components to realize the full potential of APSC performance.
Co-reporter:Michael W. Mara; David N. Bowman; Onur Buyukcakir; Megan L. Shelby; Kristoffer Haldrup; Jier Huang; Michael R. Harpham; Andrew B. Stickrath; Xiaoyi Zhang▽; J. Fraser Stoddart; Ali Coskun; Elena Jakubikova
Journal of the American Chemical Society 2015 Volume 137(Issue 30) pp:9670-9684
Publication Date(Web):July 8, 2015
DOI:10.1021/jacs.5b04612
Copper(I) diimine complexes have emerged as low cost replacements for ruthenium complexes as light sensitizers and electron donors, but their shorter metal-to-ligand-charge-transfer (MLCT) states lifetimes and lability of transient Cu(II) species impede their intended functions. Two carboxylated Cu(I) bis-2,9-diphenylphenanthroline (dpp) complexes [Cu(I)(dpp-O(CH2CH2O)5)(dpp-(COOH)2)]+ and [Cu(I)(dpp-O(CH2CH2O)5)(dpp-(Φ-COOH)2)]+ (Φ = tolyl) with different linker lengths were synthesized in which the MLCT-state solvent quenching pathways are effectively blocked, the lifetime of the singlet MLCT state is prolonged, and the transient Cu(II) ligands are stabilized. Aiming at understanding the mechanisms of structural influence to the interfacial charge transfer in the dye-sensitized solar cell mimics, electronic and geometric structures as well as dynamics for the MLCT state of these complexes and their hybrid with TiO2 nanoparticles were investigated using optical transient spectroscopy, X-ray transient absorption spectroscopy, time-dependent density functional theory, and quantum dynamics simulations. The combined results show that these complexes exhibit strong absorption throughout the visible spectrum due to the severely flattened ground state, and a long-lived charge-separated Cu(II) has been achieved via ultrafast electron injection (<300 fs) from the 1MLCT state into TiO2 nanoparticles. The results also indicate that the TiO2-phen distance in these systems does not have significant effect on the efficiency of the interfacial electron-transfer process. The mechanisms for electron transfer in these systems are discussed and used to develop new strategies in optimizing copper(I) diimine complexes in solar energy conversion devices.
Co-reporter:Alexander M. Schneider, Luyao Lu, Eric F. Manley, Tianyue Zheng, Valerii Sharapov, Tao Xu, Tobin J. Marks, Lin X. Chen and Luping Yu
Chemical Science 2015 vol. 6(Issue 8) pp:4860-4866
Publication Date(Web):04 Jun 2015
DOI:10.1039/C5SC01427A
We report the properties of a new series of wide band gap photovoltaic polymers based on the N-alkyl 2-pyridone dithiophene (PDT) unit. These polymers are effective bulk heterojunction solar cell materials when blended with phenyl-C71-butyric acid methyl ester (PC71BM). They achieve power conversion efficiencies (up to 5.33%) high for polymers having such large bandgaps, ca. 2.0 eV (optical) and 2.5 eV (electrochemical). Grazing incidence wide-angle X-ray scattering (GIWAXS) reveals strong correlations between π–π stacking distance and regularity, polymer backbone planarity, optical absorption maximum energy, and photovoltaic efficiency.
Co-reporter:Nicholas E. Jackson; Brett M. Savoie; Tobin J. Marks; Lin X. Chen;Mark A. Ratner
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 1) pp:77-84
Publication Date(Web):December 11, 2014
DOI:10.1021/jz502223t
While the intense focus on energy level tuning in organic photovoltaic materials has afforded large gains in device performance, we argue here that strategies based on microstructural/morphological control are at least as promising in any rational design strategy. In this work, a meta-analysis of ∼150 bulk heterojunction devices fabricated with different materials combinations is performed and reveals strong correlations between power conversion efficiency and morphology-dominated properties (short-circuit current, fill factor) and surprisingly weak correlations between efficiency and energy level positioning (open-circuit voltage, enthalpic offset at the interface, optical gap). While energy level positioning should in principle provide the theoretical maximum efficiency, the optimization landscape that must be navigated to reach this maximum is unforgiving. Thus, research aimed at developing understanding-based strategies for more efficient optimization of an active layer microstructure and morphology are likely to be at least as fruitful.
Co-reporter:Jangdae Youn;Sureshraju Vegiraju;Jonathan D. Emery;Benjamin J. Leever;Sumit Kewalramani;Silvia J. Lou;Shiming Zhang;Kumaresan Prabakaran;Yamuna Ezhumalai;Choongik Kim;Peng-Yi Huang;Charlotte Stern;Wen-Chung Chang;Michael J. Bedzyk;Ming-Chou Chen;Antonio Facchetti;Tobin J. Marks
Advanced Electronic Materials 2015 Volume 1( Issue 8) pp:
Publication Date(Web):
DOI:10.1002/aelm.201500098
Three new fused thiophene semiconductors, end-capped with diperfluorophenylthien-2-yl (DFPT) groups (DFPT-thieno[2′,3′:4,5]thieno[3,2-b]thieno[2,3-d]thiophene (TTA), DFPT-dithieno[2,3-b:3′,2′-d]thiophenes (DTT), and DFPT-thieno[3,2-b]thiophene (TT)), are synthesized and characterized in organic thin film transistors. Good environmental stability of the newly developed materials is demonstrated via thermal analysis as well as degradation tests under white light. The molecular structures of all three perfluorophenylthien-2-yl end-functionalized derivatives are determined by single crystal X-ray diffraction. DFPT-TTA and DFPT-TT exhibit good n-type TFT performance, with mobilities up to 0.43 and 0.33 cm2 V−1 s−1, respectively. These are among the best performing n-type materials of all fused thiophenes reported to date. The best thin film transistor device performance is achieved via an n-octadecyltrichlorosilane dielectric surface treatment on the thermally grown Si/SiO2 substrates prior to vapor-phase semiconductor deposition. Within the DFPT series, carrier mobility magnitudes depend strongly on the semiconductor growth conditions and the gate dielectric surface treatment.
Co-reporter:Nanjia Zhou;Hui Lin;Sylvia J. Lou;Xinge Yu;Peijun Guo;Eric F. Manley;Stephen Loser;Patrick Hartnett;Hui Huang;Michael R. Wasielewski;Robert P. H. Chang;Antonio Facchetti;Tobin J. Marks
Advanced Energy Materials 2014 Volume 4( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/aenm.201300785
Co-reporter:Charles Kiseok Song ; Kyle A. Luck ; Nanjia Zhou ; Li Zeng ; Henry M. Heitzer ; Eric F. Manley ; Samuel Goldman ; Lin X. Chen ; Mark A. Ratner ; Michael J. Bedzyk ; Robert P. H. Chang ; Mark C. Hersam ;Tobin J. Marks
Journal of the American Chemical Society 2014 Volume 136(Issue 51) pp:17762-17773
Publication Date(Web):November 24, 2014
DOI:10.1021/ja508453n
To achieve densely packed charge-selective organosilane-based interfacial layers (IFLs) on the tin-doped indium oxide (ITO) anodes of organic photovoltaic (OPV) cells, a series of Ar2N-(CH2)n-SiCl3 precursors with Ar = 3,4-difluorophenyl, n = 3, 6, 10, and 18, was synthesized, characterized, and chemisorbed on OPV anodes to serve as IFLs. To minimize lateral nonbonded -NAr2···Ar2N- repulsions which likely limit IFL packing densities in the resulting self-assembled monolayers (SAMs), precursor mixtures having both small and large n values are simultaneously deposited. These “heterogeneous” SAMs are characterized by a battery of techniques: contact angle measurements, X-ray reflectivity, X-ray photoelectron spectroscopy, ultraviolet photoelectron spectroscopy (UPS), cyclic voltammetry, and DFT computation. It is found that the headgroup densities of these “supersaturated” heterogeneous SAMs (SHSAMs) are enhanced by as much as 17% versus their homogeneous counterparts. Supersaturation significantly modifies the IFL properties including the work function (as much as 16%) and areal dipole moment (as much as 49%). Bulk-heterojunction OPV devices are fabricated with these SHSAMs: ITO/IFL/poly[[4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl][2-[[(2-ethylhexyl)oxy]carbonyl]-3-fluorothieno[3,4-b]thiophenediyl]]:phenyl-C71-butyric acid methyl ester (PTB7:PC71BM)/LiF/Al. OPVs having SHSAM IFLs exhibit significantly enhanced performance (PCE by 54%; Voc by 35%) due to enhanced charge selectivity and collection, with the PCE rivaling or exceeding that of PEDOT:PSS IFL devices −7.62%. The mechanism underlying the enhanced performance involves modified hole collection and selectivity efficiency inferred from the UPS data. The ITO/SAM/SHSAM surface potential imposed by the dipolar SAMs causes band bending and favorably alters the Schottky barrier height. Thus, interfacial charge selectivity and collection are enhanced as evident in the greater OPV Voc.
Co-reporter:L. X. Chen, X. Zhang and M. L. Shelby
Chemical Science 2014 vol. 5(Issue 11) pp:4136-4152
Publication Date(Web):30 Jun 2014
DOI:10.1039/C4SC01333F
As an X-ray method for capturing transient structures of molecules during chemical reactions, X-ray transient absorption (XTA), or laser-initiated time-resolved X-ray absorption spectroscopy, has seen its capabilities greatly expanded over the past decade. XTA, which includes X-ray absorption near edge structure (XANES) and X-ray absorption fine structure (XAFS), has evolved beyond proof-of-concept studies and has been increasingly used to interrogate real chemical problems. Advances in ultrafast laser technology, pulsed X-ray sources in synchrotron facilities, as well as the frontier femtosecond X-ray pulses from X-ray free electron lasers have opened up new opportunities to gain a new fundamental description of the chemical sciences. This review reports historical and recent advances in XTA, particularly in its chemical applications, and is focused on (1) an overview of XTA capabilities in comparison to the related techniques of X-ray emission spectroscopy (XES) and resonant inelastic X-ray scattering (RIXS), (2) general chemical properties that can be investigated by the XTA method, (3) chemical systems studied by XTA investigations such as transition metal complexes, metalloproteins and hybrid systems, and (4) summary and perspectives.
Co-reporter:J. Huang, M. W. Mara, A. B. Stickrath, O. Kokhan, M. R. Harpham, K. Haldrup, M. L. Shelby, X. Zhang, R. Ruppert, J.-P. Sauvage and L. X. Chen
Dalton Transactions 2014 vol. 43(Issue 47) pp:17615-17623
Publication Date(Web):10 Sep 2014
DOI:10.1039/C4DT02046D
Photophysical and structural properties of a CuI diimine complex with very strong steric hindrance, [CuI(dppS)2]+ (dppS = 2,9-diphenyl-1,10-phenanthroline disulfonic acid disodium salt), are investigated by optical and X-ray transient absorption (OTA and XTA) spectroscopy. The bulky phenylsulfonic acid groups at 2,9 positions of phenanthroline ligands force the ground state and the metal-to-ligand charge-transfer (MLCT) excited state to adopt a flattened pseudo-tetrahedral coordination geometry in which the solvent access to the copper center is completely blocked. We analyzed the MLCT state dynamics and structures as well as those of the charge separated state resulting from the interfacial electron injection from the MLCT state to TiO2 nanoparticles (NPs). The OTA results show the absence of the sub-picosecond component previously assigned as the time constant for flattening, while the two observed time constants are assigned to a relatively slow intersystem crossing (ISC) rate (∼13.8 ps) and a decay rate (100 ns) of the [CuI(dppS)2]+ MLCT state in water. These results correlate well with the XTA studies that resolved a flattened tetrahedral Cu(I) coordination geometry in the ground state. Probing the 3MLCT state structure with XTA establishes that the 3MLCT state has the same oxidation state as the copper center in [CuII(dppS)2]2+ and the Cu–N distance is reduced by 0.06 Å compared to that of the ground state, accompanied by a rotation of phenyl rings located at 2,9 positions of phenanthroline. The structural dynamics of the photoinduced charge transfer process in the [CuI(dppS)2]+/TiO2 hybrid is also investigated, which suggests a more restricted environment for the complex upon binding to TiO2 NPs. Moreover, the Cu–N bond length of the oxidized state of [CuI(dppS)2]+ after electron injection to TiO2 NPs shortens by 0.05 Å compared to that in the ground state. The interpretation of these observed structural changes associated with excited and charge separated states will be discussed. These results not only set an example for applying XTA in capturing the intermediate structure of metal complex/semiconductor NP hybrids but also provide guidance for designing efficient CuI diimine complexes with optimized structures for application in solar-to-electricity conversion.
Co-reporter:Kelly A. Fransted, Nicholas E. Jackson, Ruifa Zong, Michael W. Mara, Jier Huang, Michael R. Harpham, Megan L. Shelby, Randolph P. Thummel, and Lin X. Chen
The Journal of Physical Chemistry A 2014 Volume 118(Issue 45) pp:10497-10506
Publication Date(Web):July 11, 2014
DOI:10.1021/jp504294j
In this study, ultrafast optical transient absorption and X-ray transient absorption (XTA) spectroscopy are used to probe the excited-state dynamics and structural evolution of copper(I) bicinchoninic acid ([Cu(I)(BCA)2]+), which has similar but less frequently studied biquinoline-based ligands compared to phenanthroline-based complexes. The optical transient absorption measurements performed on the complex in a series of polar protic solvents demonstrate a strong solvent dependency for the excited lifetime, which ranges from approximately 40 ps in water to over 300 ps in 2-methoxyethanol. The XTA experiments showed a reduction of the prominent 1s → 4pz edge peak in the excited-state X-ray absorption near-edge structure (XANES) spectrum, which is indicative of an interaction with a fifth ligand, most likely the solvent. Analysis of the extended X-ray absorption fine structure (EXAFS) spectrum shows a shortening of the metal–ligand bond in the excited state and an increase in the coordination number for the Cu(II) metal center. A flattened structure is supported by DFT calculations that show that the system relaxes into a flattened geometry with a lowest-energy triplet state that has a dipole-forbidden transition to the ground state. While the short excited-state lifetime relative to previously studied Cu(I) diimine complexes could be attributed to this dark triplet state, the strong solvent dependency and the reduction of the 1s → 4pz peak in the XTA data suggest that solvent interaction could also play a role. This detailed study of the dynamics in different solvents provides guidance for modulating excited-state pathways and lifetimes through structural factors such as solvent accessibility to fulfill the excited-state property requirements for efficient light harvesting and electron injection.
Co-reporter:Tianyue Zheng, Luyao Lu, Nicholas E. Jackson, Sylvia J. Lou, Lin X. Chen, and Luping Yu
Macromolecules 2014 Volume 47(Issue 18) pp:6252-6259
Publication Date(Web):September 9, 2014
DOI:10.1021/ma501152v
This work describes an efficient synthetic method for creating ladder-type, oligomeric donor monomers with fused thienobenzothiophene structures. These monomers are copolymerized with fluorinated thieno[3,4-b]thiophene ester to form a series of polymers which are investigated as donor materials in polymer/fullerene solar cells. Photophysical and electrochemical characterizations are used in conjunction with quantum-chemical calculations to identify the interplay of quinoidal and charge transfer character in the optical gaps of conjugated copolymers, providing broadly applicable design rules for tuning the excitation character of conjugated copolymers. X-ray diffraction, mobility measurements, and solar cell device characterization are used to analyze neat films and bulk heterojunctions of these copolymers, demonstrating the importance of the spatial symmetry of the donor and acceptor unit in determining the charge transport characteristics of conjugated copolymers.
Co-reporter:Nicholas E. Jackson ; Brett M. Savoie ; Kevin L. Kohlstedt ; Monica Olvera de la Cruz ; George C. Schatz ; Lin X. Chen ;Mark A. Ratner
Journal of the American Chemical Society 2013 Volume 135(Issue 28) pp:10475-10483
Publication Date(Web):June 25, 2013
DOI:10.1021/ja403667s
The chemical variety present in the organic electronics literature has motivated us to investigate potential nonbonding interactions often incorporated into conformational “locking” schemes. We examine a variety of potential interactions, including oxygen–sulfur, nitrogen–sulfur, and fluorine–sulfur, using accurate quantum-chemical wave function methods and noncovalent interaction (NCI) analysis on a selection of high-performing conjugated polymers and small molecules found in the literature. In addition, we evaluate a set of nonbonding interactions occurring between various heterocyclic and pendant atoms taken from a group of representative π-conjugated molecules. Together with our survey and set of interactions, it is determined that while many nonbonding interactions possess weak binding capabilities, nontraditional hydrogen-bonding interactions, oxygen–hydrogen (CH···O) and nitrogen–hydrogen (CH···N), are alone in inducing conformational control and enhanced planarity along a polymer or small molecule backbone at room temperature.
Co-reporter:Charles Kiseok Song, Alicia C. White, Li Zeng, Benjamin J. Leever, Michael D. Clark, Jonathan D. Emery, Sylvia J. Lou, Amod Timalsina, Lin X. Chen, Michael J. Bedzyk, and Tobin J. Marks
ACS Applied Materials & Interfaces 2013 Volume 5(Issue 18) pp:9224
Publication Date(Web):August 13, 2013
DOI:10.1021/am4030609
With the goal of investigating and enhancing anode performance in bulk-heterojunction (BHJ) organic photovoltaic (OPV) cells, the glass/tin-doped indium oxide (ITO) anodes are modified with a series of robust silane-tethered bis(fluoroaryl)amines to form self-assembled interfacial layers (IFLs). The modified ITO anodes are characterized by contact angle measurements, X-ray reflectivity, ultraviolet photoelectron spectroscopy, X-ray photoelectron spectroscopy, grazing incidence X-ray diffraction, atomic force microscopy, and cyclic voltammetry. These techniques reveal the presence of hydrophobic amorphous monolayers of 6.68 to 9.76 Å thickness, and modified anode work functions ranging from 4.66 to 5.27 eV. Two series of glass/ITO/IFL/active layer/LiF/Al BHJ OPVs are fabricated with the active layer = poly(3-hexylthiophene):phenyl-C71-butyric acid methyl ester (P3HT:PC71BM) or poly[[4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b’]dithiophene-2,6-diyl][3-fluoro-2-[(2-ethylhexyl)-carbonyl]thi-eno[3,4-b]thiophenediyl]]:phenyl-C71-butyric acid methyl ester (PTB7:PC71BM). OPV analysis under AM 1.5G conditions reveals significant performance enhancement versus unmodified glass/ITO anodes. Strong positive correlations between the electrochemically derived heterogeneous electron transport rate constants (ks) and the device open circuit voltage (Voc), short circuit current (Jsc), hence OPV power conversion efficiency (PCE), are observed for these modified anodes. Furthermore, the strong functional dependence of the device response on ks increases as greater densities of charge carriers are generated in the BHJ OPV active layer, and is attributable to enhanced anode carrier extraction in the case of high-ks IFLs.Keywords: heterogeneous electron transfer rate constant; open circuit voltage; organic photovoltaics; power conversion efficiency; self-assembled monolayer; work function;
Co-reporter:Michael W. Mara, Nicholas E. Jackson, Jier Huang, Andrew B. Stickrath, Xiaoyi Zhang, Nosheen A. Gothard, Mark A. Ratner, and Lin X. Chen
The Journal of Physical Chemistry B 2013 Volume 117(Issue 6) pp:1921-1931
Publication Date(Web):January 16, 2013
DOI:10.1021/jp311643t
The effects of structural constraints on the metal-to-ligand charge transfer (MLCT) excited state structural dynamics of cuprous bis-2,9-diphenyl-phenanthroline ([Cu(I)(dpp)2]+) in both coordinating acetonitrile and noncoordinating toluene were studied using X-ray transient absorption (XTA) spectroscopy and density functional theory (DFT) calculations. The phenyl groups attached to the phenanthroline ligands not only effectively shield the Cu(I) center from solvent molecules, but also force a flattened tetrahedral coordination geometry of the Cu(I) center. Consequently, the MLCT state lifetime in [Cu(I)(dpp)2]+ is solvent-independent, unlike the previously studied 2,9-methyl substituted bis-phenanthroline Cu(I) complex. The MLCT state of [Cu(I)(dpp)2]+ still undergoes a “pseudo Jahn-Teller distortion,” with the angle between the two phenanthroline ligand planes decreased further by 7°. The XTA results indicate that, in the MLCT excited state of [Cu(I)(dpp)2]+, the phenyls at the 2, 9 positions of the phenanthroline rotate, breaking the π–π interaction with the phenanthroline ligands without ever rotating in-plane with the phenanthroline ligands. Hence, the transferred electron density from the Cu(I) center is localized on the phenanthroline moiety with no charge density present on the phenyl rings. The insight about the effect of the structural constraints on the MLCT state properties will guide the design of Cu(I) diimine complexes with suitable excited-state properties to function as earth-abundant dye sensitizers for solar electricity generation.
Co-reporter:Andrew B. Stickrath, Michael W. Mara, Jenny V. Lockard, Michael R. Harpham, Jier Huang, Xiaoyi Zhang, Klaus Attenkofer, and Lin X. Chen
The Journal of Physical Chemistry B 2013 Volume 117(Issue 16) pp:4705-4712
Publication Date(Web):November 15, 2012
DOI:10.1021/jp3086705
Although understanding the structural dynamics associated with ligand photodissociation is necessary in order to correlate structure and function in biological systems, few techniques are capable of measuring the ultrafast dynamics of these systems in solution-phase at room temperature. We present here a detailed X-ray transient absorption (XTA) study of the photodissociation of CO-bound myoglobin (Fe(II)CO-Mb) in room-temperature aqueous buffer solution with a time resolution of 80 ps, along with a general procedure for handling biological samples under the harsh experimental conditions that transient X-ray experiments entail. The XTA spectra of (Fe(II)CO-Mb) exhibit significant XANES and XAFS alterations following 527 nm excitation, which remain unchanged for >47 μs. These spectral changes indicate loss of the CO ligand, resulting in a five-coordinate, domed heme, and significant energetic reorganization of the 3d orbitals of the Fe center. With the current experimental setup, each X-ray pulse in the pulse train, separated by ∼153 ns, can be separately discriminated, yielding snapshots of the myoglobin evolution over time. These methods can be easily applied to other biological systems, allowing for simultaneous structural and electronic measurements of any biological system with both ultrafast and slow time resolutions, effectively mapping out all of the samples’ relevant physiological processes.
Co-reporter:Nosheen A. Gothard, Michael W. Mara, Jier Huang, Jodi M. Szarko, Brian Rolczynski, Jenny V. Lockard, and Lin X. Chen
The Journal of Physical Chemistry A 2012 Volume 116(Issue 9) pp:1984-1992
Publication Date(Web):January 31, 2012
DOI:10.1021/jp211646p
The metal-to-ligand-charge-transfer (MLCT) excited state of Cu(I) diimine complexes is known to undergo structural reorganization, transforming from a pseudotetrahedral D2d symmetry in the ground state to a flattened D2 symmetry in the MLCT state, which allows ligation with a solvent molecule, forming an exciplex intermediate. Therefore, the structural factors that influence the coordination geometry change and the solvent accessibility to the copper center in the MLCT state could be used to control the excited state properties. In this study, we investigated an extreme case of the steric hindrance caused by attaching bulky tert-butyl groups in bis(2,9-di-tert-butyl-1,10-phenanthroline)copper(I), [CuI(dtbp)2]+. The two bulky tert-butyl groups on the dtbp ligand lock the MLCT state into the pseudotetrahedral coordination geometry and completely block the solvent access to the copper center in the MLCT state of [CuI(dtbp)2]+. Using ultrafast transient absorption spectroscopy and time-resolved emission spectroscopy, we investigated the MLCT state property changes due to the steric hindrance and demonstrated that [CuI(dtbp)2]+ exhibited a long-lived emission but no subpicosecond component that was previously assigned as the flattening of the pseudotetrahedral coordination geometry. This suggests the retention of its pseudotetrahedral D2d symmetry and the blockage of the solvent accessibility. We made a comparison between the excited state dynamics of [CuI(dtbp)2]+ with its mono-tert-butyl counterpart, bis(2-tert-butyl-1,10-phenanthroline)copper(I) [CuI(tbp)2]+. The subpicosecond component assigned to the flattening of the D2d coordination geometry in the MLCT excited state was again present in the latter because the absence of a tert-butyl on the phenanthroline allows flattening to the pseudotetrahedral coordination geometry. Unlike the [CuI(dtbp)2]+, [CuI(tbp)2]+ exhibited no detectable emission at room temperature in solution. These results provide new insights into the manipulation of various excited state properties in Cu diimine complexes by certain key structural factors, enabling optimization of these systems for solar energy conversion applications.
Co-reporter:Sylvia J. Lou ; Jodi M. Szarko ; Tao Xu ; Luping Yu ; Tobin J. Marks
Journal of the American Chemical Society 2011 Volume 133(Issue 51) pp:20661-20663
Publication Date(Web):November 29, 2011
DOI:10.1021/ja2085564
Processing additives are used in organic photovoltaic systems to optimize the active layer film morphology. However, the actual mechanism is not well understood. Using X-ray scattering techniques, we analyze the effects of an additive diiodooctane (DIO) on the aggregation of a high-efficiency donor polymer PTB7 and an acceptor molecule PC71BM under solar cell processing conditions. We conclude that DIO selectively dissolves PC71BM aggregates, allowing their intercalation into PTB7 domains, thereby optimizing both the domain size and the PTB7–PC71BM interface.
Co-reporter:Jodi M. Szarko, Jianchang Guo, Brian S. Rolczynski and Lin X. Chen
Journal of Materials Chemistry A 2011 vol. 21(Issue 22) pp:7849-7857
Publication Date(Web):07 Apr 2011
DOI:10.1039/C0JM04433D
Applications of low band gap polymers in solar cells have attracted intense attention due to their energetic overlap with the solar spectrum. Recently, low band gap organic photovoltaic (OPV) materials have shown an unprecedented ∼8% efficiency in solar cell devices. Although the energetic alignment is crucial in the optimization of these materials, the structural and kinetic effects are also important factors in the overall device performance. Here we focus on the morphology and charge separation kinetics of several energetically similar low band gap materials. Special emphasis will be on two polymers, PF and PTB, in this report.
Co-reporter:Jianchang Guo, Yongye Liang, Jodi Szarko, Byeongdu Lee, Hae Jung Son, Brian S. Rolczynski, Luping Yu and Lin X. Chen
The Journal of Physical Chemistry B 2010 Volume 114(Issue 2) pp:742-748
Publication Date(Web):December 28, 2009
DOI:10.1021/jp909135k
Molecular packing structures and photoinduced charge separation dynamics have been investigated in a recently developed bulk heterojunction (BHJ) organic photovoltaic (OPV) material based on poly(thienothiophene-benzodithiophene) (PTB1) with a power conversion efficiency (PCE) of >5% in solar cell devices. Grazing incidence X-ray scattering (GIXS) measurements of the PTB1:PCBM ([6,6]-phenyl-C61-butyric acid methyl ester) films revealed π-stacked polymer backbone planes oriented parallel to the substrate surface, in contrast to the π-stacked polymer backbone planes oriented perpendicular to the substrate surface in regioregular P3HT [poly(3-hexylthiophene)]:PCBM films. A ∼1.7 times higher charge mobility in the PTB1:PCBM film relative to that in P3HT:PCBM films is attributed to this difference in stacking orientation. The photoinduced charge separation (CS) rate in the pristine PTB1:PCBM film is more than twice as fast as that in the annealed P3HT:PCBM film. The combination of a small optical gap, fast CS rate, and high carrier mobility in the PTB1:PCBM film contributes to its relatively high PCE in the solar cells. Contrary to P3HT:PCBM solar cells, annealing PTB1:PCBM films reduced the device PCE from 5.24% in the pristine film to 1.92% due to reduced interfacial area between the electron donor and the acceptor. Consequently, quantum yields of exciton generation and charge separation in the annealed film are significantly reduced compared to those in the pristine film.
Co-reporter:Alexander M. Schneider, Luyao Lu, Eric F. Manley, Tianyue Zheng, Valerii Sharapov, Tao Xu, Tobin J. Marks, Lin X. Chen and Luping Yu
Chemical Science (2010-Present) 2015 - vol. 6(Issue 8) pp:NaN4866-4866
Publication Date(Web):2015/06/04
DOI:10.1039/C5SC01427A
We report the properties of a new series of wide band gap photovoltaic polymers based on the N-alkyl 2-pyridone dithiophene (PDT) unit. These polymers are effective bulk heterojunction solar cell materials when blended with phenyl-C71-butyric acid methyl ester (PC71BM). They achieve power conversion efficiencies (up to 5.33%) high for polymers having such large bandgaps, ca. 2.0 eV (optical) and 2.5 eV (electrochemical). Grazing incidence wide-angle X-ray scattering (GIWAXS) reveals strong correlations between π–π stacking distance and regularity, polymer backbone planarity, optical absorption maximum energy, and photovoltaic efficiency.
Co-reporter:L. X. Chen, X. Zhang and M. L. Shelby
Chemical Science (2010-Present) 2014 - vol. 5(Issue 11) pp:NaN4152-4152
Publication Date(Web):2014/06/30
DOI:10.1039/C4SC01333F
As an X-ray method for capturing transient structures of molecules during chemical reactions, X-ray transient absorption (XTA), or laser-initiated time-resolved X-ray absorption spectroscopy, has seen its capabilities greatly expanded over the past decade. XTA, which includes X-ray absorption near edge structure (XANES) and X-ray absorption fine structure (XAFS), has evolved beyond proof-of-concept studies and has been increasingly used to interrogate real chemical problems. Advances in ultrafast laser technology, pulsed X-ray sources in synchrotron facilities, as well as the frontier femtosecond X-ray pulses from X-ray free electron lasers have opened up new opportunities to gain a new fundamental description of the chemical sciences. This review reports historical and recent advances in XTA, particularly in its chemical applications, and is focused on (1) an overview of XTA capabilities in comparison to the related techniques of X-ray emission spectroscopy (XES) and resonant inelastic X-ray scattering (RIXS), (2) general chemical properties that can be investigated by the XTA method, (3) chemical systems studied by XTA investigations such as transition metal complexes, metalloproteins and hybrid systems, and (4) summary and perspectives.
Co-reporter:Jodi M. Szarko, Jianchang Guo, Brian S. Rolczynski and Lin X. Chen
Journal of Materials Chemistry A 2011 - vol. 21(Issue 22) pp:NaN7857-7857
Publication Date(Web):2011/04/07
DOI:10.1039/C0JM04433D
Applications of low band gap polymers in solar cells have attracted intense attention due to their energetic overlap with the solar spectrum. Recently, low band gap organic photovoltaic (OPV) materials have shown an unprecedented ∼8% efficiency in solar cell devices. Although the energetic alignment is crucial in the optimization of these materials, the structural and kinetic effects are also important factors in the overall device performance. Here we focus on the morphology and charge separation kinetics of several energetically similar low band gap materials. Special emphasis will be on two polymers, PF and PTB, in this report.
Co-reporter:J. Huang, M. W. Mara, A. B. Stickrath, O. Kokhan, M. R. Harpham, K. Haldrup, M. L. Shelby, X. Zhang, R. Ruppert, J.-P. Sauvage and L. X. Chen
Dalton Transactions 2014 - vol. 43(Issue 47) pp:NaN17623-17623
Publication Date(Web):2014/09/10
DOI:10.1039/C4DT02046D
Photophysical and structural properties of a CuI diimine complex with very strong steric hindrance, [CuI(dppS)2]+ (dppS = 2,9-diphenyl-1,10-phenanthroline disulfonic acid disodium salt), are investigated by optical and X-ray transient absorption (OTA and XTA) spectroscopy. The bulky phenylsulfonic acid groups at 2,9 positions of phenanthroline ligands force the ground state and the metal-to-ligand charge-transfer (MLCT) excited state to adopt a flattened pseudo-tetrahedral coordination geometry in which the solvent access to the copper center is completely blocked. We analyzed the MLCT state dynamics and structures as well as those of the charge separated state resulting from the interfacial electron injection from the MLCT state to TiO2 nanoparticles (NPs). The OTA results show the absence of the sub-picosecond component previously assigned as the time constant for flattening, while the two observed time constants are assigned to a relatively slow intersystem crossing (ISC) rate (∼13.8 ps) and a decay rate (100 ns) of the [CuI(dppS)2]+ MLCT state in water. These results correlate well with the XTA studies that resolved a flattened tetrahedral Cu(I) coordination geometry in the ground state. Probing the 3MLCT state structure with XTA establishes that the 3MLCT state has the same oxidation state as the copper center in [CuII(dppS)2]2+ and the Cu–N distance is reduced by 0.06 Å compared to that of the ground state, accompanied by a rotation of phenyl rings located at 2,9 positions of phenanthroline. The structural dynamics of the photoinduced charge transfer process in the [CuI(dppS)2]+/TiO2 hybrid is also investigated, which suggests a more restricted environment for the complex upon binding to TiO2 NPs. Moreover, the Cu–N bond length of the oxidized state of [CuI(dppS)2]+ after electron injection to TiO2 NPs shortens by 0.05 Å compared to that in the ground state. The interpretation of these observed structural changes associated with excited and charge separated states will be discussed. These results not only set an example for applying XTA in capturing the intermediate structure of metal complex/semiconductor NP hybrids but also provide guidance for designing efficient CuI diimine complexes with optimized structures for application in solar-to-electricity conversion.

![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3-(5-bromo-2-thienyl)-2,5-bis(2-ethylhexyl)-2,5-dihydro-6-(2-thienyl)-](/data/chemimg/139400/1308671-90-0.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3-(5-bromo-2-thienyl)-2,5-bis(2-ethylhexyl)-2,5-dihydro-6-(2-thienyl)-](/data/chemimg/139400/1308671-90-0_b.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3,6-bis(5-bromo-2-thienyl)-2,5-dihydro-2,5-bis(2-octyldodecyl)-](/data/chemimg/139000/1260685-63-9.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3,6-bis(5-bromo-2-thienyl)-2,5-dihydro-2,5-bis(2-octyldodecyl)-](/data/chemimg/139000/1260685-63-9_b.png)
![Thieno[3,4-b]thiophene-2-carboxylic acid, 4,6-dibromo-3-fluoro-, 2-ethylhexyl ester](/data/chemimg/1642600/1237479-38-7.png)
![Thieno[3,4-b]thiophene-2-carboxylic acid, 4,6-dibromo-3-fluoro-, 2-ethylhexyl ester](/data/chemimg/1642600/1237479-38-7_b.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3,6-bis(5-bromo-2-thienyl)-2,5-bis(2-butyloctyl)-2,5-dihydro-](/data/chemimg/2260700/1224709-68-5.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3,6-bis(5-bromo-2-thienyl)-2,5-bis(2-butyloctyl)-2,5-dihydro-](/data/chemimg/2260700/1224709-68-5_b.png)


![2-Ethylhexyl 4,6-dibromo-3-fluorothieno[3,4-b]thiophene-2-carboxylate](http://img.cochemist.com/ccimg/1237500/1237479-39-8.png)
![2-Ethylhexyl 4,6-dibromo-3-fluorothieno[3,4-b]thiophene-2-carboxylate](http://img.cochemist.com/ccimg/1237500/1237479-39-8_b.png)
![2,5-Bis(2-ethylhexyl)-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione](http://img.cochemist.com/ccimg/1185900/1185885-86-2.png)
![2,5-Bis(2-ethylhexyl)-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione](http://img.cochemist.com/ccimg/1185900/1185885-86-2_b.png)



![2,5-Bis(trimethylstannyl)thieno[3,2-b]thiophene](http://img.cochemist.com/ccimg/470000/469912-82-1.png)
![2,5-Bis(trimethylstannyl)thieno[3,2-b]thiophene](http://img.cochemist.com/ccimg/470000/469912-82-1_b.png)


![Poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene]](http://img.cochemist.com/ccimg/138200/138184-36-8.png)
![Poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene]](http://img.cochemist.com/ccimg/138200/138184-36-8_b.png)


![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(1-pentylhexyl)-](/data/chemimg/3689300/110590-83-5.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-bis(1-pentylhexyl)-](/data/chemimg/3689300/110590-83-5_b.png)







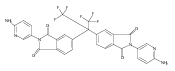
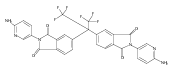
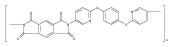
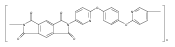


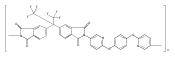
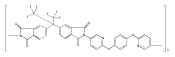


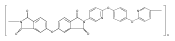
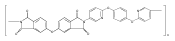














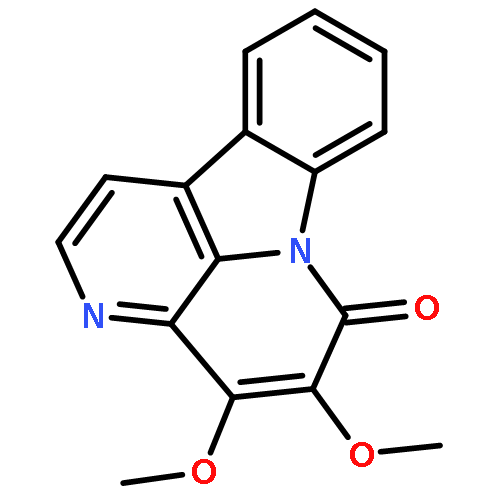
![2,3-Dihydro-3-methyl-6H-indolo[3,2,1-de][1,5]naphthyridine-2,6-dione](http://img.cochemist.com/ccimg/82700/82652-21-9.png)
![2,3-Dihydro-3-methyl-6H-indolo[3,2,1-de][1,5]naphthyridine-2,6-dione](http://img.cochemist.com/ccimg/82700/82652-21-9_b.png)
![Pyridine, 2,2'-[1,4-phenylenebis(oxy)]bis[5-nitro-](http://img.cochemist.com/ccimg/53400/53304-64-6.png)
![Pyridine, 2,2'-[1,4-phenylenebis(oxy)]bis[5-nitro-](http://img.cochemist.com/ccimg/53400/53304-64-6_b.png)
![POLY[(1,3-DIHYDRO-1,3-DIOXO-2H-ISOINDOLE-2,5-DIYL)CARBONYL(1,3-DIHYDRO-1,3-DIOXO-2H-ISOINDOLE-5,2-DIYL)-5,2-PYRIDINEDIYLOXY-1,4-PHENYLENEOXY-2,5-PYRIDINEDIYL]](http://img.cochemist.com/ccimg/53400/53301-84-1.png)
![POLY[(1,3-DIHYDRO-1,3-DIOXO-2H-ISOINDOLE-2,5-DIYL)CARBONYL(1,3-DIHYDRO-1,3-DIOXO-2H-ISOINDOLE-5,2-DIYL)-5,2-PYRIDINEDIYLOXY-1,4-PHENYLENEOXY-2,5-PYRIDINEDIYL]](http://img.cochemist.com/ccimg/53400/53301-84-1_b.png)

![5,7-dihydroxy-2-(4-hydroxy-3-methoxyphenyl)-3-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-[[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxychromen-4-one](http://img.cochemist.com/ccimg/17500/17429-69-5.png)
![5,7-dihydroxy-2-(4-hydroxy-3-methoxyphenyl)-3-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-[[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxychromen-4-one](http://img.cochemist.com/ccimg/17500/17429-69-5_b.png)