Co-reporter:Shuguang Chen, WeiJun Zhou, Qing Zhang, YanHo Kwok, GuanHua Chen, and Mark A. Ratner
The Journal of Physical Chemistry Letters October 19, 2017 Volume 8(Issue 20) pp:5166-5166
Publication Date(Web):October 4, 2017
DOI:10.1021/acs.jpclett.7b02214
Quantum interference in cross-conjugated molecules can be utilized to construct molecular quantum interference effect transistors. However, whether its application can be achieved depends on the survivability of the quantum interference under real conditions such as nuclear vibration. We use two simulation methods to investigate the effects of nuclear vibration on quantum interference in a meta-linked benzene system. The simulation results suggest that the quantum interference is robust against nuclear vibration not only in the steady state but also in its transient dynamics, and thus the molecular quantum interference effect transistors can be realized.
Co-reporter:Martín A. Mosquera, Nicholas E. Jackson, Thomas J. Fauvell, Matthew S. Kelley, Lin X. Chen, George C. Schatz, and Mark A. Ratner
Journal of the American Chemical Society March 15, 2017 Volume 139(Issue 10) pp:3728-3728
Publication Date(Web):February 22, 2017
DOI:10.1021/jacs.6b12405
The theoretical description of the time-evolution of excitons requires, as an initial step, the calculation of their spectra, which has been inaccessible to most users due to the high computational scaling of conventional algorithms and accuracy issues caused by common density functionals. Previously (J. Chem. Phys. 2016, 144, 204105), we developed a simple method that resolves these issues. Our scheme is based on a two-step calculation in which a linear-response TDDFT calculation is used to generate orbitals perturbed by the excitonic state, and then a second linear-response TDDFT calculation is used to determine the spectrum of excitations relative to the excitonic state. Herein, we apply this theory to study near-infrared absorption spectra of excitons in oligomers of the ubiquitous conjugated polymers poly(3-hexylthiophene) (P3HT), poly(2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene) (MEH-PPV), and poly(benzodithiophene-thieno[3,4-b]thiophene) (PTB7). For P3HT and MEH-PPV oligomers, the calculated intense absorption bands converge at the longest wavelengths for 10 monomer units, and show strong consistency with experimental measurements. The calculations confirm that the exciton spectral features in MEH-PPV overlap with those of the bipolaron formation. In addition, our calculations identify the exciton absorption bands in transient absorption spectra measured by our group for oligomers (1, 2, and 3 units) of PTB7. For all of the cases studied, we report the dominant orbital excitations contributing to the optically active excited state–excited state transitions, and suggest a simple rule to identify absorption peaks at the longest wavelengths. We suggest our methodology could be considered for further developments in theoretical transient spectroscopy to include nonadiabatic effects, coherences, and to describe the formation of species such as charge-transfer states and polaron pairs.
Co-reporter:Colin Van Dyck, Tobin J. Marks, and Mark A. Ratner
ACS Nano June 27, 2017 Volume 11(Issue 6) pp:5970-5970
Publication Date(Web):June 2, 2017
DOI:10.1021/acsnano.7b01807
Dielectric materials are ubiquitous in optics, electronics, and materials science. Recently, there have been new efforts to characterize the dielectric performance of thin films composed of molecular assemblies. In this context, we investigate here the relationship between the polarizability of the constituent molecules and the film dielectric constant, using periodic density functional theory (DFT) calculations, for polyyne and saturated alkane chains. In particular, we explore the implication of the superlinear chain length dependence of the polarizability, a specific feature of conjugated molecules. We show and explain from DFT calculations and a simple depolarization model that this superlinearity is attenuated by the collective polarization. However, it is not completely suppressed. This confers a very high sensitivity of the dielectric constant to the thin film thickness. This latter can increase by a factor of 3–4 at reasonable coverages, by extending the molecular length. This significantly limits the decline of the thin film capacitance with the film thickness. Therefore, the conventional fit of the capacitance versus thickness is not appropriate to determine the dielectric constant of the film. Finally, we show that the failures of semilocal approximations of the exchange-correlation functional lead to a very significant overestimation of this effect.Keywords: chain length; conjugation; depolarization; dielectric constant; permittivity; thickness; thin film;
Co-reporter:Daniel D. Powell, Michael R. Wasielewski, and Mark A. Ratner
The Journal of Physical Chemistry B July 27, 2017 Volume 121(Issue 29) pp:7190-7190
Publication Date(Web):June 29, 2017
DOI:10.1021/acs.jpcb.7b02748
Coherence effects on electron transfer in a series of symmetric and asymmetric two-, three-, four-, and five-site molecular model systems for photosystem I in cyanobacteria and green plants were studied. The total site energies of the electronic Hamiltonian were calculated using the density functional theory (DFT) formalism and included the zero point vibrational energies of the electron donors and acceptors. Site energies and couplings were calculated using a polarizable continuum model to represent various solvent environments, and the site-to-site couplings were calculated using fragment charge difference methods at the DFT level of theory. The Redfield formalism was used to propagate the electron density from the donors to the acceptors, incorporating relaxation and dephasing effects to describe the electron transfer processes. Changing the relative energies of the donor, intermediate acceptor, and final acceptor molecules in these assemblies has profound effects on the electron transfer rates as well as on the amplitude of the quantum oscillations observed. Increasing the ratio of a particular energy gap to the electronic coupling for a given pair of states leads to weaker quantum oscillations between sites. Biasing the intermediate acceptor energies to slightly favor one pathway leads to a general decrease in electron transfer yield.
Co-reporter:Brett M. Savoie, Akshay Rao, Artem A. Bakulin, Simon Gelinas, Bijan Movaghar, Richard H. Friend, Tobin J. Marks, and Mark A. Ratner
Journal of the American Chemical Society February 19, 2014 Volume 136(Issue 7) pp:2876-2884
Publication Date(Web):January 24, 2014
DOI:10.1021/ja411859m
Natural photosynthetic complexes accomplish the rapid conversion of photoexcitations into spatially separated electrons and holes through precise hierarchical ordering of chromophores and redox centers. In contrast, organic photovoltaic (OPV) cells are poorly ordered, utilize only two different chemical potentials, and the same materials that absorb light must also transport charge; yet, some OPV blends achieve near-perfect quantum efficiency. Here we perform electronic structure calculations on large clusters of functionalized fullerenes of different size and ordering, predicting several features of the charge generation process, outside the framework of conventional theories but clearly observed in ultrafast electro-optical experiments described herein. We show that it is the resonant coupling of photogenerated singlet excitons to a high-energy manifold of fullerene electronic states that enables efficient charge generation, bypassing localized charge-transfer states. In contrast to conventional views, our findings suggest that fullerene cluster size, concentration, and dimensionality control charge generation efficiency, independent of exciton delocalization.
Co-reporter:Colin Van DyckMark A. Ratner
The Journal of Physical Chemistry C 2017 Volume 121(Issue 5) pp:
Publication Date(Web):January 17, 2017
DOI:10.1021/acs.jpcc.6b07855
Single-molecule junctions are the constitutive components of molecular electronics circuits. For any potential application, the energy gap in the junction, i.e., the accumulated energy difference between the electrode Fermi level and the two frontier energy levels of the molecule, is a key property. Here, using the nonequilibrium Green’s function coupled to the density functional theory framework (NEGF-DFT) method, we show that the gap of the molecule inserted between electrodes can differ largely from the gap of the same molecule, at the isolated level. It can be widely compressed by tuning the alignment mechanism at each metal/molecule interface. In the context of molecular rectification, we show that this mechanism relates to the pinning effect. We discuss the different parameters affecting the compression of the gap and its efficiency. Interestingly, we find that the structure both of the molecule and of the anchoring group plays an important role. Finally, we investigate the evolution of these features out-of-equilibrium.
Co-reporter:Henry M. Heitzer, Tobin J. Marks, and Mark A. Ratner
Accounts of Chemical Research 2016 Volume 49(Issue 9) pp:1614
Publication Date(Web):August 30, 2016
DOI:10.1021/acs.accounts.6b00173
The dielectric response of a material is central to numerous processes spanning the fields of chemistry, materials science, biology, and physics. Despite this broad importance across these disciplines, describing the dielectric environment of a molecular system at the level of first-principles theory and computation remains a great challenge and is of importance to understand the behavior of existing systems as well as to guide the design and synthetic realization of new ones. Furthermore, with recent advances in molecular electronics, nanotechnology, and molecular biology, it has become necessary to predict the dielectric properties of molecular systems that are often difficult or impossible to measure experimentally. In these scenarios, it is would be highly desirable to be able to determine dielectric response through efficient, accurate, and chemically informative calculations.A good example of where theoretical modeling of dielectric response would be valuable is in the development of high-capacitance organic gate dielectrics for unconventional electronics such as those that could be fabricated by high-throughput printing techniques. Gate dielectrics are fundamental components of all transistor-based logic circuitry, and the combination high dielectric constant and nanoscopic thickness (i.e., high capacitance) is essential to achieving high switching speeds and low power consumption. Molecule-based dielectrics offer the promise of cheap, flexible, and mass producible electronics when used in conjunction with unconventional organic or inorganic semiconducting materials to fabricate organic field effect transistors (OFETs). The molecular dielectrics developed to date typically have limited dielectric response, which results in low capacitances, translating into poor performance of the resulting OFETs. Furthermore, the development of better performing dielectric materials has been hindered by the current highly empirical and labor-intensive pace of synthetic progress. An accurate and efficient theoretical computational approach could drastically decrease this time by screening potential dielectric materials and providing reliable design rules for future molecular dielectrics.Until recently, accurate calculation of dielectric responses in molecular materials was difficult and highly approximate. Most previous modeling efforts relied on classical formalisms to relate molecular polarizability to macroscopic dielectric properties. These efforts often vastly overestimated polarizability in the subject materials and ignored crucial material properties that can affect dielectric response. Recent advances in first-principles calculations via density functional theory (DFT) with periodic boundary conditions have allowed accurate computation of dielectric properties in molecular materials.In this Account, we outline the methodology used to calculate dielectric properties of molecular materials. We demonstrate the validity of this approach on model systems, capturing the frequency dependence of the dielectric response and achieving quantitative accuracy compared with experiment. This method is then used as a guide to new high-capacitance molecular dielectrics by determining what materials and chemical properties are important in maximizing dielectric response in self-assembled monolayers (SAMs). It will be seen that this technique is a powerful tool for understanding and designing new molecular dielectric systems, the properties of which are fundamental to many scientific areas.
Co-reporter:Yang Yang, Martín A. Mosquera, Kwan Skinner, Andres E. Becerra, Vasgen Shamamian, George C. Schatz, Mark A. Ratner, and Tobin J. Marks
The Journal of Physical Chemistry A 2016 Volume 120(Issue 47) pp:9476-9488
Publication Date(Web):November 2, 2016
DOI:10.1021/acs.jpca.6b09526
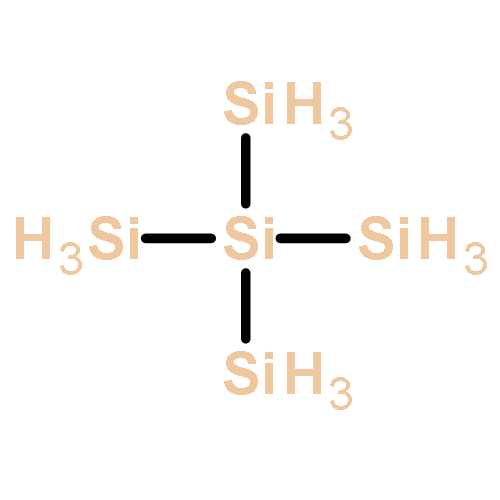
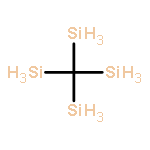
Silicon-based materials are crucial for conventional electronics. The fascinating properties of the new two-dimensional material silicene, the silicon analogue of graphene (one atom-thick silicon sheets), offer a potential bridge between conventional and molecular electronics. The ground-state configuration of silicene is buckled, which compromises optimal constructive overlap of p orbitals. Because silicene is not planar like graphene, it has a lower intrinsic electron/hole mobility than graphene. This motivates a search for improved, alternative, planar materials. Miniaturization of silicene/graphene hybrid monolayers affords diverse silicon-organic and -inorganic molecules, whose potential as building blocks for molecular electronics is unexplored. Additionally, hybridization of pure silicon rings (or sheets) is a versatile way to control the geometrical and electronic characteristics of the aromatic ring. In this work we systematically investigate, computationally, architectures and electronic structures of a series of hybrid silaaromatic monomers and fused-ring oligomers. This includes the thermochemistry of representative reactions: hydrogenation and oxidation. The effect of various skeletal substituents of interest is elucidated as well. We show that the specific location of carbon and silicon atoms, and their relative populations in the rings are crucial factors controlling the molecular geometry and the quasi-particle gap. Furthermore, we suggest that electron-withdrawing substituents such as CN, F, and CF3 are promising candidates to promote the air-stability of silaaromatics. Finally, on the basis of the analysis of benzene-like silaaromatic molecules, we discuss a set of alternative, prototype ring molecules that feature planarity and delocalized π bonds. These motifs may be useful for designing new extended materials.
Co-reporter:Amrit Poudel; Xin Chen
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 6) pp:955-960
Publication Date(Web):February 25, 2016
DOI:10.1021/acs.jpclett.6b00119
The high density of evanescent modes in the vicinity of a metal leads to enhancement of the near-field Förster resonant energy transfer (FRET) rate. We present a classical approach to calculate the FRET rate based on the dyadic Green’s function of an arbitrary dielectric environment and consider the nonlocal limit of material permittivity in the case of the metallic half-space and thin film. In a dimer system, we find that the FRET rate is enhanced due to shared evanescent photon modes bridging a donor and an acceptor. Furthermore, a general expression for the FRET rate for multimer systems is derived. The presence of a dielectric environment and the path interference effect enhance the transfer rate, depending on the combination of distance and geometry.
Co-reporter:Brett M. Savoie;Scott Dunaisky;Tobin J. Marks
Advanced Energy Materials 2015 Volume 5( Issue 3) pp:
Publication Date(Web):
DOI:10.1002/aenm.201400891
Co-reporter:Colin Van Dyck and Mark A. Ratner
Nano Letters 2015 Volume 15(Issue 3) pp:1577-1584
Publication Date(Web):February 23, 2015
DOI:10.1021/nl504091v
The quest for a molecular rectifier is among the major challenges of molecular electronics. We introduce three simple rules to design an efficient rectifying molecule and demonstrate its functioning at the theoretical level, relying on the NEGF-DFT technique. The design rules notably require both the introduction of asymmetric anchoring moieties and a decoupling bridge. They lead to a new rectification mechanism based on the compression and control of the HOMO/LUMO gap by the electrode Fermi levels, arising from a pinning effect. Significant rectification ratios up to 2 orders of magnitude are theoretically predicted as the mechanism opposes resonant to nonresonant tunneling.
Co-reporter:Melanie R. Butler, Bijan Movaghar, Tobin J. Marks, and Mark A. Ratner
Nano Letters 2015 Volume 15(Issue 3) pp:1597-1602
Publication Date(Web):January 23, 2015
DOI:10.1021/nl5041176
We propose a set of design rules with a model Hamiltonian that allows electrons to form attracting pairs through the exploitation of a new combination of resonant band alignment and Coulombic repulsion. The pair bands and single particle bands in various lattices are calculated and compared in energy, and regions of net attraction are identified. This work provides guidelines for the construction of molecular systems, nanocrystals, and nanoparticle arrays with the potential for superconductivity.
Co-reporter:Nicholas E. Jackson; Kevin L. Kohlstedt; Brett M. Savoie; Monica Olvera de la Cruz; George C. Schatz; Lin X. Chen
Journal of the American Chemical Society 2015 Volume 137(Issue 19) pp:6254-6262
Publication Date(Web):April 28, 2015
DOI:10.1021/jacs.5b00493
With the abundant variety and increasing chemical complexity of conjugated polymers proliferating the field of organic semiconductors, it has become increasingly important to correlate the polymer molecular structure with its mesoscale conformational and morphological attributes. For instance, it is unknown which combinations of chemical moieties and periodicities predictably produce mesoscale ordering. Interestingly, not all ordered morphologies result in efficient devices. In this work we have parametrized accurate classical force-fields and used these to compute the conformational and aggregation characteristics of single strands of common conjugated polymers. Molecular dynamics trajectories are shown to reproduce experimentally observed polymeric ordering, concluding that efficient organic photovoltaic devices span a range of polymer conformational classes, and suggesting that the solution-phase morphologies have far-reaching effects. Encouragingly, these simulations indicate that despite the wide-range of conformational classes present in successful devices, local molecular ordering, and not long-range crystallinity, appears to be the necessary requirement for efficient devices. Finally, we examine what makes a “good” solvent for conjugated polymers, concluding that dispersive π-electron solvent–polymer interactions, and not the electrostatic potential of the backbone interacting with the solvent, are what primarily determine a polymer’s solubility in a particular solvent, and consequently its morphological characteristics.
Co-reporter:Henry M. Heitzer; Tobin J. Marks
Journal of the American Chemical Society 2015 Volume 137(Issue 22) pp:7189-7196
Publication Date(Web):May 15, 2015
DOI:10.1021/jacs.5b03301
Donor–bridge–acceptor (DBA) systems occupy a rich history in molecular electronics and photonics. A key property of DBA materials is their typically large and tunable (hyper)polarizabilities. While traditionally, classical descriptions such as the Clausius–Mossotti formalism have been used to relate molecular polarizabilities to bulk dielectric response, recent work has shown that these classical equations are inadequate for numerous materials classes. Creating high-dielectric organic materials is critically important for utilizing unconventional semiconductors in electronic circuitry. Employing a plane-wave density functional theory formalism, we investigate the dielectric response of highly polarizable DBA molecule-based thin films. Such films are found to have large dielectric response arising from cooperative effects between donor and acceptor units when mediated by a conjugated bridge. Moreover, the dielectric response can be systematically tuned by altering the building block donor, acceptor, or bridge structures and is found to be nonlinearly dependent on electric field strength. The computed dielectric constants are largely independent of the density functional employed, and qualitative trends are readily evident. Remarkably large computed dielectric constants >15.0 and capacitances >6.0 μF/cm2 are achieved for squaraine monolayers, significantly higher than in traditional organic dielectrics. Such calculations should provide a guide for designing high-capacitance organic dielectrics that should greatly enhance transistor performance.
Co-reporter:Nicholas E. Jackson; Brett M. Savoie; Lin X. Chen
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 6) pp:1018-1021
Publication Date(Web):March 3, 2015
DOI:10.1021/acs.jpclett.5b00135
While advances in quantum chemistry have rendered the accurate prediction of band alignment relatively straightforward, the ability to forecast a noncrystalline, multimolecule system’s conductivity possesses no simple computational form. Adapting the theory of classical resistor networks, we develop an index for quantifying charge transport in bulk molecular materials, without the requirement of crystallinity. The basic behavior of this index is illustrated through its application to simple lattices and clusters of common organic photovoltaic molecules, where it is shown to reproduce experimentally known performances for these materials. This development provides a quantitative computational means for determining a priori the bulk charge transport properties of molecular materials.
Co-reporter:Justin P. Bergfield, Henry M. Heitzer, Colin Van Dyck, Tobin J. Marks, and Mark A. Ratner
ACS Nano 2015 Volume 9(Issue 6) pp:6412
Publication Date(Web):May 26, 2015
DOI:10.1021/acsnano.5b02042
We investigate the relationship between dielectric response and charge transport in molecule-based materials operating in the quantum coherent regime. We find that quantum interference affects these observables differently, for instance, allowing current passing through certain materials to be reduced by orders of magnitude without affecting dielectric behavior (or band gap). As an example, we utilize ab initio electronic structure theory to calculate conductance and dielectric constants of cross-conjugated anthraquinone (AQ)-based and linearly conjugated anthracene (AC)-based materials. In spite of having nearly equal fundamental gaps, electrode bonding configurations, and molecular dimensions, we find a ∼1.7 order of magnitude (∼50-fold) reduction in the conductance of the AQ-based material relative to the AC-based material, a value in close agreement with recent measurements, while the calculated dielectric constants of both materials are nearly identical. From these findings, we propose two molecular materials in which quantum interference is used to reduce leakage currents across a ∼25 Å monolayer gap with dielectric constants larger than 4.5.Keywords: cross-conjugated polymers; density functional theory; molecular dielectric material; nonequilibrium quantum transport; quantum interference;
Co-reporter:Brad S. Veldkamp, Xinle Liu, Michael R. Wasielewski, Joseph E. Subotnik, and Mark A. Ratner
The Journal of Physical Chemistry A 2015 Volume 119(Issue 2) pp:253-262
Publication Date(Web):October 22, 2014
DOI:10.1021/jp508337x
We show for a series of six small donor–acceptor dyads that the energy difference between non-charge transfer (non-CT) and charge transfer (CT) excited states, as well as the squares of the electronic couplings between these states, can be predicted from first-principles using variational orbital adapted configuration interaction singles (VOA-CIS) theory. VOA-CIS correctly predicts the observed experimental trends in these values and provides quantitative accuracy roughly on par with a modern long-range corrected density functional, ωB97X. Using VOA-CIS and ωB97X, the experimental energy difference between the non-CT and CT excited states is predicted with root mean squared errors of 0.22 eV and 0.21 eV, respectively. The square of the electronic coupling between these states is predicted with root mean squared errors of 0.08 eV2 and 0.07 eV2, respectively. Orbital optimized CIS (OO-CIS) and CIS(D), two perturbative corrections to CIS, provide a significant correction to the errant relative energies predicted by CIS, but the correction is insufficient to recover the experimentally observed trend.
Co-reporter:Nicholas E. Jackson; Brett M. Savoie; Tobin J. Marks; Lin X. Chen
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 1) pp:77-84
Publication Date(Web):December 11, 2014
DOI:10.1021/jz502223t
While the intense focus on energy level tuning in organic photovoltaic materials has afforded large gains in device performance, we argue here that strategies based on microstructural/morphological control are at least as promising in any rational design strategy. In this work, a meta-analysis of ∼150 bulk heterojunction devices fabricated with different materials combinations is performed and reveals strong correlations between power conversion efficiency and morphology-dominated properties (short-circuit current, fill factor) and surprisingly weak correlations between efficiency and energy level positioning (open-circuit voltage, enthalpic offset at the interface, optical gap). While energy level positioning should in principle provide the theoretical maximum efficiency, the optimization landscape that must be navigated to reach this maximum is unforgiving. Thus, research aimed at developing understanding-based strategies for more efficient optimization of an active layer microstructure and morphology are likely to be at least as fruitful.
Co-reporter:Zhihai Li, Manuel Smeu, Tae-Hong Park, Jeff Rawson, Yangjun Xing, Michael J. Therien, Mark A. Ratner, and Eric Borguet
Nano Letters 2014 Volume 14(Issue 10) pp:5493-5499
Publication Date(Web):September 25, 2014
DOI:10.1021/nl502466a
Single molecule break junction experiments and nonequilibrium Green’s function calculations using density functional theory (NEGF-DFT) of carbodithioate- and thiol-terminated [5,15-bis(phenylethynyl)-10,20-diarylporphinato]zinc(II) complexes reveal the impact of the electrode-linker coordination mode on charge transport at the single-molecule level. Replacement of thiolate (−S–) by the carbodithioate (−CS2–) anchoring motif leads to an order of magnitude increase of single molecule conductance. In contrast to thiolate-terminated structures, metal–molecule–metal junctions that exploit the carbodithioate linker manifest three distinct conductance values. We hypothesize that the magnitudes of these conductances depend upon carbodithoate linker hapticity with measured conductances across Au-[5,15-bis(4′-(dithiocarboxylate)phenylethynyl)-10,20-diarylporphinato]zinc(II)-Au junctions the greatest when both anchoring groups attach to the metal surface in a bidentate fashion. We support this hypothesis with NEGF-DFT calculations, which consider the electron transport properties for specific binding geometries. These results provide new insights into the origin of molecule-to-molecule conductance heterogeneity in molecular charge transport measurements and the factors that optimize electrode–molecule–electrode electronic coupling and maximize the conductance for charge transport.
Co-reporter:Charles Kiseok Song ; Kyle A. Luck ; Nanjia Zhou ; Li Zeng ; Henry M. Heitzer ; Eric F. Manley ; Samuel Goldman ; Lin X. Chen ; Mark A. Ratner ; Michael J. Bedzyk ; Robert P. H. Chang ; Mark C. Hersam ;Tobin J. Marks
Journal of the American Chemical Society 2014 Volume 136(Issue 51) pp:17762-17773
Publication Date(Web):November 24, 2014
DOI:10.1021/ja508453n
To achieve densely packed charge-selective organosilane-based interfacial layers (IFLs) on the tin-doped indium oxide (ITO) anodes of organic photovoltaic (OPV) cells, a series of Ar2N-(CH2)n-SiCl3 precursors with Ar = 3,4-difluorophenyl, n = 3, 6, 10, and 18, was synthesized, characterized, and chemisorbed on OPV anodes to serve as IFLs. To minimize lateral nonbonded -NAr2···Ar2N- repulsions which likely limit IFL packing densities in the resulting self-assembled monolayers (SAMs), precursor mixtures having both small and large n values are simultaneously deposited. These “heterogeneous” SAMs are characterized by a battery of techniques: contact angle measurements, X-ray reflectivity, X-ray photoelectron spectroscopy, ultraviolet photoelectron spectroscopy (UPS), cyclic voltammetry, and DFT computation. It is found that the headgroup densities of these “supersaturated” heterogeneous SAMs (SHSAMs) are enhanced by as much as 17% versus their homogeneous counterparts. Supersaturation significantly modifies the IFL properties including the work function (as much as 16%) and areal dipole moment (as much as 49%). Bulk-heterojunction OPV devices are fabricated with these SHSAMs: ITO/IFL/poly[[4,8-bis[(2-ethylhexyl)oxy]benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl][2-[[(2-ethylhexyl)oxy]carbonyl]-3-fluorothieno[3,4-b]thiophenediyl]]:phenyl-C71-butyric acid methyl ester (PTB7:PC71BM)/LiF/Al. OPVs having SHSAM IFLs exhibit significantly enhanced performance (PCE by 54%; Voc by 35%) due to enhanced charge selectivity and collection, with the PCE rivaling or exceeding that of PEDOT:PSS IFL devices −7.62%. The mechanism underlying the enhanced performance involves modified hole collection and selectivity efficiency inferred from the UPS data. The ITO/SAM/SHSAM surface potential imposed by the dipolar SAMs causes band bending and favorably alters the Schottky barrier height. Thus, interfacial charge selectivity and collection are enhanced as evident in the greater OPV Voc.
Co-reporter:Nicholas E. Jackson, Brett M. Savoie, Kevin L. Kohlstedt, Tobin J. Marks, Lin X. Chen, and Mark A. Ratner
Macromolecules 2014 Volume 47(Issue 3) pp:987-992
Publication Date(Web):January 21, 2014
DOI:10.1021/ma4023923
Quantum-chemical computation is a useful and widespread tool for understanding the electronic structure of conjugated polymers as well as predicting new synthetic targets. In this work, we assess the validity of considering a single conformational or structural isomer as representative of the entire conformational or structural distributions in ab-initio computations of figures-of-merit (dipole moment, HOMO, LUMO, and optical gap). It is found from surveying numerous conjugated copolymers that considering only a single conformational or structural isomer can hide significant deviations in frontier molecular orbital energies and optical gaps as well as qualitative shifts in dipole moments. We discuss the limitations of not considering isomeric dispersion on the polymer’s computed electronic properties and the implications these findings have on the rational design of conjugated polymers.
Co-reporter:Dr. Zhihai Li;Dr. Manuel Smeu;Sepideh Afsari;Dr. Yangjun Xing; Mark A. Ratner; Eric Borguet
Angewandte Chemie International Edition 2014 Volume 53( Issue 4) pp:1098-1102
Publication Date(Web):
DOI:10.1002/anie.201308398
Abstract
Sensors play a significant role in the detection of toxic species and explosives, and in the remote control of chemical processes. In this work, we report a single-molecule-based pH switch/sensor that exploits the sensitivity of dye molecules to environmental pH to build metal–molecule–metal (m-M-m) devices using the scanning tunneling microscopy (STM) break junction technique. Dyes undergo pH-induced electronic modulation due to reversible structural transformation between a conjugated and a nonconjugated form, resulting in a change in the HOMO–LUMO gap. The dye-mediated m-M-m devices react to environmental pH with a high on/off ratio (≈100:1) of device conductivity. Density functional theory (DFT) calculations, carried out under the non-equilibrium Green’s function (NEGF) framework, model charge transport through these molecules in the two possible forms and confirm that the HOMO–LUMO gap of dyes is nearly twice as large in the nonconjugated form as in the conjugated form.
Co-reporter:Henry M. Heitzer, Tobin J. Marks, and Mark A. Ratner
ACS Nano 2014 Volume 8(Issue 12) pp:12587
Publication Date(Web):November 21, 2014
DOI:10.1021/nn505431p
Developing high-capacitance organic gate dielectrics is critical for advances in electronic circuitry based on unconventional semiconductors. While high-dielectric constant molecular substances are known, the mechanism of dielectric response and the fundamental chemical design principles are not well understood. Using a plane-wave density functional theory formalism, we show that it is possible to map the atomic-scale dielectric profiles of molecule-based materials while capturing important bulk characteristics. For molecular films, this approach reveals how basic materials properties such as surface coverage density, molecular tilt angle, and π-system planarity can dramatically influence dielectric response. Additionally, relatively modest molecular backbone and substituent variations can be employed to substantially enhance film dielectric response. For dense surface coverages and proper molecular alignment, conjugated hydrocarbon chains can achieve dielectric constants of >8.0, more than 3 times that of analogous saturated chains, ∼2.5. However, this conjugation-related dielectric enhancement depends on proper molecular orientation and planarization, with enhancements up to 60% for proper molecular alignment with the applied field and an additional 30% for conformations such as coplanarity in extended π-systems. Conjugation length is not the only determinant of dielectric response, and appended polarizable high-Z substituents can increase molecular film response more than 2-fold, affording estimated capacitances of >9.0 μF/cm2. However, in large π-systems, polar substituent effects are substantially attenuated.Keywords: density functional theory; dielectric computation; field-effect transistor; organic dielectric film; self-assembled monolayer;
Co-reporter:Brett M. Savoie;Kevin L. Kohlstedt;Nicholas E. Jackson;Lin X. Chen;Monica Olvera de la Cruz;George C. Schatz;Tobin J. Marks
PNAS 2014 Volume 111 (Issue 28 ) pp:10055-10060
Publication Date(Web):2014-07-15
DOI:10.1073/pnas.1409514111
High-performance solution-processed organic semiconductors maintain macroscopic functionality even in the presence of microscopic
disorder. Here we show that the functional robustness of certain organic materials arises from the ability of molecules to
create connected mesoscopic electrical networks, even in the absence of periodic order. The hierarchical network structures
of two families of important organic photovoltaic acceptors, functionalized fullerenes and perylene diimides, are analyzed
using a newly developed graph methodology. The results establish a connection between network robustness and molecular topology,
and also demonstrate that solubilizing moieties play a large role in disrupting the molecular networks responsible for charge
transport. A clear link is established between the success of mono and bis functionalized fullerene acceptors in organic photovoltaics
and their ability to construct mesoscopically connected electrical networks over length scales of 10 nm.
Co-reporter:ShuGuang Chen, Yu Zhang, SiuKong Koo, Heng Tian, ChiYung Yam, GuanHua Chen, and Mark A. Ratner
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 15) pp:2748-2752
Publication Date(Web):June 3, 2014
DOI:10.1021/jz5007143
The primary issue in molecular electronics is measuring and understanding how electrons travel through a single molecule strung between two electrodes. A key area involves electronic interference that occurs when electrons can follow more than one pathway through the molecular entity. When the phases developed along parallel pathways are inequivalent, interference effects can substantially reduce overall conductance. This fundamentally interesting issue can be understood using classical rules of physical organic chemistry, and the subject has been examined broadly. However, there has been little dynamical study of such interference effects. Here, we use the simplest electronic structure model to examine the coherent time-dependent transport through meta- and para-linked benzene circuits, and the effects of decoherence. We find that the phase-caused coherence/decoherence behavior is established very quickly (femtoseconds), that the localized dephasing at any site reduces the destructive interference of the meta-linked species (raising the conductance), and that thermal effects are essentially ineffectual for removing coherence effects.Keywords: decoherence; molecular electronics; quantum interference; quantum transport; transient current; transistor;
Co-reporter:Dr. Zhihai Li;Dr. Manuel Smeu;Sepideh Afsari;Dr. Yangjun Xing; Mark A. Ratner; Eric Borguet
Angewandte Chemie 2014 Volume 126( Issue 4) pp:1116-1120
Publication Date(Web):
DOI:10.1002/ange.201308398
Abstract
Sensors play a significant role in the detection of toxic species and explosives, and in the remote control of chemical processes. In this work, we report a single-molecule-based pH switch/sensor that exploits the sensitivity of dye molecules to environmental pH to build metal–molecule–metal (m-M-m) devices using the scanning tunneling microscopy (STM) break junction technique. Dyes undergo pH-induced electronic modulation due to reversible structural transformation between a conjugated and a nonconjugated form, resulting in a change in the HOMO–LUMO gap. The dye-mediated m-M-m devices react to environmental pH with a high on/off ratio (≈100:1) of device conductivity. Density functional theory (DFT) calculations, carried out under the non-equilibrium Green’s function (NEGF) framework, model charge transport through these molecules in the two possible forms and confirm that the HOMO–LUMO gap of dyes is nearly twice as large in the nonconjugated form as in the conjugated form.
Co-reporter:Nicholas E. Jackson, Lin X. Chen, and Mark A. Ratner
The Journal of Physical Chemistry B 2014 Volume 118(Issue 19) pp:5194-5202
Publication Date(Web):April 28, 2014
DOI:10.1021/jp5024197
Using a combination of classical molecular dynamics and symmetry adapted intermolecular perturbation theory, we develop a high-accuracy computational method for examining the solubility energetics of nonelectrolytes. This approach is used to accurately compute the cohesive energy density and Hildebrand solubility parameters of 26 molecular liquids. The energy decomposition of symmetry adapted perturbation theory is then utilized to develop multicomponent Hansen-like solubility parameters. These parameters are shown to reproduce the solvent categorizations (nonpolar, polar aprotic, or polar protic) of all molecular liquids studied while lending quantitative rigor to these qualitative categorizations via the introduction of simple, easily computable parameters. Notably, we find that by monitoring the first-order exchange energy contribution to the total interaction energy, one can rigorously determine the hydrogen bonding character of a molecular liquid. Finally, this method is applied to compute explicitly the Flory interaction parameter and the free energy of mixing for two different small molecule mixtures, reproducing the known miscibilities. This methodology represents an important step toward the prediction of molecular solubility from first principles.
Co-reporter:Henry M. Heitzer;Brett M. Savoie; Tobin J. Marks; Mark A. Ratner
Angewandte Chemie International Edition 2014 Volume 53( Issue 29) pp:7456-7460
Publication Date(Web):
DOI:10.1002/anie.201402568
Abstract
Organic photovoltaics (OPVs) offer the opportunity for cheap, lightweight and mass-producible devices. However, an incomplete understanding of the charge generation process, in particular the timescale of dynamics and role of exciton diffusion, has slowed further progress in the field. We report a new Kinetic Monte Carlo model for the exciton dissociation mechanism in OPVs that addresses the origin of ultra-fast (<1 ps) dissociation by incorporating exciton delocalization. The model reproduces experimental results, such as the diminished rapid dissociation with increasing domain size, and also lends insight into the interplay between mixed domains, domain geometry, and exciton delocalization. Additionally, the model addresses the recent dispute on the origin of ultra-fast exciton dissociation by comparing the effects of exciton delocalization and impure domains on the photo-dynamics.This model provides insight into exciton dynamics that can advance our understanding of OPV structure–function relationships.
Co-reporter:Henry M. Heitzer;Brett M. Savoie; Tobin J. Marks; Mark A. Ratner
Angewandte Chemie International Edition 2014 Volume 53( Issue 29) pp:
Publication Date(Web):
DOI:10.1002/anie.201405393
Co-reporter:Henry M. Heitzer ; Tobin J. Marks
Journal of the American Chemical Society 2013 Volume 135(Issue 26) pp:9753-9759
Publication Date(Web):June 4, 2013
DOI:10.1021/ja401904d
The dielectric properties of materials are of fundamental significance to many chemical processes and the functioning of numerous solid-state device technologies. While experimental methods for measuring bulk dielectric constants are well-established, far less is known, either experimentally or theoretically, about the origin of dielectric response at the molecular/multimolecular scale. In this contribution we report the implementation of an accurate first-principles approach to calculating the dielectric response of molecular systems. We assess the accuracy of the method by reproducing the experimental dielectric constants of several bulk π-electron materials and demonstrating the ability of the method to capture dielectric properties as a function of frequency and molecular orientation in representative arrays of substituted aromatic derivatives. The role of molecular alignment and packing density on dielectric response is also examined, showing that the local dielectric behavior of molecular assemblies can diverge significantly from that of the bulk material.
Co-reporter:Nicholas E. Jackson ; Brett M. Savoie ; Kevin L. Kohlstedt ; Monica Olvera de la Cruz ; George C. Schatz ; Lin X. Chen
Journal of the American Chemical Society 2013 Volume 135(Issue 28) pp:10475-10483
Publication Date(Web):June 25, 2013
DOI:10.1021/ja403667s
The chemical variety present in the organic electronics literature has motivated us to investigate potential nonbonding interactions often incorporated into conformational “locking” schemes. We examine a variety of potential interactions, including oxygen–sulfur, nitrogen–sulfur, and fluorine–sulfur, using accurate quantum-chemical wave function methods and noncovalent interaction (NCI) analysis on a selection of high-performing conjugated polymers and small molecules found in the literature. In addition, we evaluate a set of nonbonding interactions occurring between various heterocyclic and pendant atoms taken from a group of representative π-conjugated molecules. Together with our survey and set of interactions, it is determined that while many nonbonding interactions possess weak binding capabilities, nontraditional hydrogen-bonding interactions, oxygen–hydrogen (CH···O) and nitrogen–hydrogen (CH···N), are alone in inducing conformational control and enhanced planarity along a polymer or small molecule backbone at room temperature.
Co-reporter:Lauren M. Matosziuk ; Jonathan H. Leibowitz ; Marie C. Heffern ; Keith W. MacRenaris ; Mark A. Ratner ;Thomas J. Meade
Inorganic Chemistry 2013 Volume 52(Issue 21) pp:12250-12261
Publication Date(Web):June 18, 2013
DOI:10.1021/ic400681j
We report the structural optimization and mechanistic investigation of a series of bioactivated magnetic resonance imaging contrast agents that transform from low relaxivity to high relaxivity in the presence of Zn(II). The change in relaxivity results from a structural transformation of the complex that alters the coordination environment about the Gd(III) center. Here, we have performed a series of systematic modifications to determine the structure that provides the optimal change in relaxivity in response to the presence of Zn(II). Relaxivity measurements in the presence and absence of Zn(II) were used in conjunction with measurements regarding water access (namely, number of water molecules bound) to the Gd(III) center and temperature-dependent 13C NMR spectroscopy to determine how the coordination environment about the Gd(III) center is affected by the distance between the Zn(II)-binding domain and the Gd(III) chelate, the number of functional groups on the Zn(II)-binding domain, and the presence of Zn(II). The results of this study provide valuable insight into the design principles for future bioactivated magnetic resonance probes.
Co-reporter:Lauren M. Matosziuk, Robert J. Holbrook, Lisa M. Manus, Marie C. Heffern, Mark A. Ratner and Thomas J. Meade
Dalton Transactions 2013 vol. 42(Issue 11) pp:4002-4012
Publication Date(Web):22 Jan 2013
DOI:10.1039/C2DT32565A
Cobalt(III) Schiff base complexes, such as [Co(acacen)L2]+, inhibit the function of Zn(II)-dependent proteins through dissociative exchange of the axial ligands with key histidine residues of the target protein. Consequently the efficacy of these compounds depends strongly on the lability of the axial ligands. A series of [Co(acacen)L2]+ complexes with various axial ligands was investigated using DFT to determine the kinetics and thermodynamics of ligand exchange and hydrolysis. Results showed excellent agreement with experimental data, indicating that axial ligand lability is determined by several factors: pKa of the axial ligand, the kinetic barrier to ligand dissociation, and the relative thermodynamic stability of the complexes before and after exchange. Hammett plots were constructed to determine if the kinetics and thermodynamics of exchange can be modulated by the addition of an electron-withdrawing group (EWG) to either the axial ligand itself or to the equatorial acacen ligand. Results predict that addition of an EWG to the axial ligand will shift the kinetics and thermodynamics so as to promote axial ligand exchange, while addition of an EWG to acacen will decrease axial ligand lability. These investigations will aid in the design of the next generation of [Co(acacen)L2]2+, allowing researchers to develop new, more effective inhibitors.
Co-reporter:Brett M. Savoie, Nicholas E. Jackson, Tobin J. Marks and Mark A. Ratner
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 13) pp:4538-4547
Publication Date(Web):01 Feb 2013
DOI:10.1039/C3CP50438G
We present results showing that common approximations employed in the design and characterization of organic photovoltaic (OPV) materials can lead to significant errors in widely adopted design rules. First, we assess the validity of the common practice of using HOMO and LUMO energies in place of formal redox potentials to characterize organic semiconductors. We trace the formal justification for this practice and survey its limits in a way that should be useful for those entering the field. We find that while the HOMO and LUMO energies represent useful descriptive approximations, they are too quantitatively inaccurate for predictive material design. Second, we show that the excitonic nature of common organic semiconductors makes it paramount to distinguish between the optical and electronic bandgaps for materials design. Our analysis shows that the usefulness of the “LUMO–LUMO Offset” as a design parameter for exciton dissociation is directly tied to the accuracy of the one-electron approximation. In particular, our results suggest that the use of the “LUMO–LUMO Offset” as a measure of the driving force for exciton dissociation leads to a systematic overestimation that should be cautiously avoided.
Co-reporter:Guangqi Li ; Bijan Movaghar
The Journal of Physical Chemistry C 2013 Volume 117(Issue 2) pp:850-857
Publication Date(Web):December 9, 2012
DOI:10.1021/jp310557h
Two simple quantum electron networks are considered: one has an interference structure, and one is a simple chain. The network is coupled at one edge site to a metal reservoir that works as a sink for arriving charges. When the electron reaches the edge site and has an energy at or above the Fermi level of the metal sink, we assume that it will be absorbed. The adiabatic electron phonon coupling will lower the energy level of the last site (before the sink) when the electron enters it. When this polarization-corrected energy is lower than the Fermi level, the absorption into the sink has to be activated above the metal Fermi sea and the absorbing rate will be slowed down.
Co-reporter:Zhihai Li, Manuel Smeu, Mark A. Ratner, and Eric Borguet
The Journal of Physical Chemistry C 2013 Volume 117(Issue 29) pp:14890-14898
Publication Date(Web):July 11, 2013
DOI:10.1021/jp309871d
Controlling charge transport through individual molecules and further understanding the effect of anchoring groups on charge transport are central themes in molecule-based devices. However, in most anchoring effect studies, only two, or at most three nonthiol anchoring groups were studied and compared for a specific system, i.e., using the same core structure. The scarcity of direct comparison data makes it difficult to draw unambiguous conclusions on the anchoring group effect. In this contribution, we focus on the single molecule conductance of porphyrins terminated with a range of anchoring groups: sulfonate (−SO3–), hydroxyl (−OH), nitrile (−CN), amine (−NH2), carboxylic acid (−COOH), benzyl (−C6H6), and pyridyl (−C6H5N). The present study represents a first attempt to investigate a broad series of anchoring groups in one specific molecule for a direct comparison. It also is the first attempt, to our knowledge, to explore single molecule conductivity with two novel anchoring groups sulfonate (−SO3–) and hydroxyl (−OH). Our experimental results reveal that the single molecule conductance values of the porphyrins follow the sequence of pyridyl > amine > sulfonate > nitrile > carboxylic acid. Electron transport calculations are in agreement that the pyridyl groups result in higher conductance values than the other groups, which is due to a stronger binding interaction of this group to the Au electrodes. The finding of a general trend in the effect of anchoring groups and the exploration of new anchoring groups reported in this paper may provide useful information for molecule-based devices, functional porphyrin design, and electron transfer/transport studies.
Co-reporter:Jonathan D. Servaites, Brett M. Savoie, Joseph B. Brink, Tobin J. Marks and Mark A. Ratner
Energy & Environmental Science 2012 vol. 5(Issue 8) pp:8343-8350
Publication Date(Web):01 May 2012
DOI:10.1039/C2EE21376A
We propose a model for geminate electron–hole dissociation in organic photovoltaic (OPV) cells and show how power conversion efficiencies greater than those currently achieved might be realized via design strategies employing moderate optical bandgaps and enhanced charge delocalization near the donor–acceptor interface. Applying this model to describing geminate electron–hole dissociation via charge transfer (CT) states, we find good agreement with recently published high-efficiency experimental data. The optimal bandgap for current-generation organic active layer materials is argued to be ∼1.7 eV – significantly greater than in previous analyses, including the Shockley–Queisser approach based upon non-excitonic solar cell dynamics. For future higher efficiency OPVs, the present results show that the optimal bandgap should be slightly lower, ∼1.6 eV. Finally, these results support design strategies aimed at enhancing mobility near the donor–acceptor interface and reducing the electron–hole binding energy, rather than striving to further reduce the bandgap.
Co-reporter:Lisa A. Fredin;Zhong Li;Michael T. Lanagan;Tobin J. Marks
Advanced Materials 2012 Volume 24( Issue 44) pp:
Publication Date(Web):
DOI:10.1002/adma.201290280
Co-reporter:Lisa A. Fredin;Zhong Li;Michael T. Lanagan;Tobin J. Marks
Advanced Materials 2012 Volume 24( Issue 44) pp:5946-5953
Publication Date(Web):
DOI:10.1002/adma.201202183
Co-reporter:Guangqi Li, Abraham Nitzan and Mark A. Ratner
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 41) pp:14270-14276
Publication Date(Web):10 Aug 2012
DOI:10.1039/C2CP41532A
A simple model is constructed to describe dissociation of charge transfer excitons in bulk heterojunction solar cells, and its dependence on the physical parameters of the system. In bulk heterojunction organic photovoltaics (OPVs), exciton dissociation occurs almost exclusively at the interface between the donor and acceptor, following one-electron initial excitation from the HOMO to the LUMO levels of the donor, and charge transfer to the acceptor to make a charge-transfer exciton. After exciton breakup, and neglecting the trapping of individual carriers, the electron may undergo two processes for decay: one process involves the electron and/or hole leaving the interface, and migrating to the electrode. This is treated here as the electron moving on a set of acceptor sites. The second loss process is radiationless decay following recombination of the acceptor electron with the donor cation; this is treated by adding a relaxation term. These two processes compete with one another. We model both the exciton breakup and the subsequent electron motion. Results depend on tunneling amplitude, energetics, disorder, Coulomb barriers, and energy level matchups, particularly the so-called LUMO–LUMO offset.
Co-reporter:Yuri A. Berlin, Alexander A. Voityuk, and Mark A. Ratner
ACS Nano 2012 Volume 6(Issue 9) pp:8216
Publication Date(Web):August 18, 2012
DOI:10.1021/nn3030139
We report a computational search for DNA π-stack structures exhibiting high electric conductance in the hopping regime, based on the INDO/S calculations of electronic coupling and the method of data analysis called k-means clustering. Using homogeneous poly(G)–poly(C) and poly(A)–poly(T) stacks as the simplest structural models, we identify the configurations of neighboring G:C and A:T pairs that allow strong electronic coupling and, therefore, molecular electric conductance much larger than the values reported for the corresponding reference systems in the literature. A computational approach for modeling the impact of thermal fluctuations on the averaged dimer structure was also proposed and applied to the [(G:C),(G:C)] and [(A:T),(A:T)] duplexes. The results of this work may provide guidance for the construction of DNA devices and DNA-based elements of nanoscale molecular circuits. Several factors that cause changes of step parameters favorable to the formation of the predicted stack conformation with high electric conductance of DNA molecules are also discussed; favorable geometries may enhance the conductivity by factors as large as 15.Keywords: DNA; electronic coupling; molecular conductance; step parameters; π-stack
Co-reporter:Michael D. Irwin, Jonathan D. Servaites, D. Bruce Buchholz, Benjamin J. Leever, Jun Liu, Jonathan D. Emery, Ming Zhang, Jung-Hwan Song, Michael F. Durstock, Arthur J. Freeman, Michael J. Bedzyk, Mark C. Hersam, Robert P. H. Chang, Mark A. Ratner, and Tobin J. Marks
Chemistry of Materials 2011 Volume 23(Issue 8) pp:2218
Publication Date(Web):April 1, 2011
DOI:10.1021/cm200229e
The functionality of NiO interfacial layers in enhancing bulk heterojunction (BHJ) organic photovoltaic (OPV) cell performance is investigated by integrated characterization of the electrical properties, microstructure, electronic structure, and optical properties of thin NiO films grown on glass/ITO electrodes. These NiO layers are found to be advantageous in BHJ OPV applications due to favorable energy band levels, interface passivation, p-type character, crystallinity, smooth surfaces, and optical transparency. The NiO overlayers are fabricated via pulsed-laser deposition and found to have a work function of ∼5.3 eV. They are investigated by both topographic and conductive atomic force microscopy and shown to passivate interfacial charge traps. The films also have an average optical transparency of >80% in the visible range, crucial for efficient OPV function, and have a near-stoichiometric Ni:O surface composition. By grazing-incidence X-ray diffraction, the NiO thin films are shown to grow preferentially in the (111) direction and to have the fcc NaCl crystal structure. Diodes of p−n structure and first-principles electronic structure calculations indicate that the NiO interlayer is preferentially conductive to holes, with a lower hole charge carrier effective mass versus that of electrons. Finally, the implications of these attributes in advancing efficiencies for state-of-the-art OPV systems—in particular, improving the open circuit voltage (VOC)—are discussed.Keywords: electron blocking layer; hole transport layer; interfacial layer; nickel oxide; NiO; organic photovoltaics; organic solar cells;
Co-reporter:N. Renaud, M. A. Ratner, and C. Joachim
The Journal of Physical Chemistry B 2011 Volume 115(Issue 18) pp:5582-5592
Publication Date(Web):February 16, 2011
DOI:10.1021/jp111384d
We present a simple method to compute the transmission coefficient of a quantum system embedded between two conducting electrodes. Starting from the solution of the time-dependent Schrodinger equation, we demonstrate the relationship between the temporal evolution of the state vector, |ψ(t)⟩, initially localized on one electrode and the electronic transmission coefficient, T(E). We particularly emphasize the role of the oscillation frequency and the decay rate of |ψ(t)⟩ in the line shape of T(E). This method is applied to the well-known problems of the single impurity, two-site systems and the benzene ring, where it agrees with well-accepted time-independent methods and gives new physical insight to the resonance and interference patterns widely observed in molecular junctions.
Co-reporter:Matthew R. Hartings, Igor V. Kurnikov, Alexander R. Dunn, Jay R. Winkler, Harry B. Gray, Mark A. Ratner
Coordination Chemistry Reviews 2010 Volume 254(3–4) pp:248-253
Publication Date(Web):February 2010
DOI:10.1016/j.ccr.2009.08.008
We report a quantitative theoretical analysis of long-range electron transfer through sensitizer wires bound in the active-site channel of cytochrome P450cam. Each sensitizer wire consists of a substrate group with high binding affinity for the enzyme active site connected to a ruthenium-diimine through a bridging aliphatic or aromatic chain. Experiments have revealed a dramatic dependence of electron transfer rates on the chemical composition of both the bridging group and the substrate. Using combined molecular dynamics simulations and electronic coupling calculations, we show that electron tunneling through perfluorinated aromatic bridges is promoted by enhanced superexchange coupling through virtual reduced states. In contrast, electron flow through aliphatic bridges occurs by hole-mediated superexchange. We have found that a small number of wire conformations with strong donor–acceptor couplings can account for the observed electron tunneling rates for sensitizer wires terminated with either ethylbenzene or adamantane. In these instances, the rate is dependent not only on electronic coupling of the donor and acceptor but also on the nuclear motion of the sensitizer wire, necessitating the calculation of average rates over the course of a molecular dynamics simulation. These calculations along with related recent findings have made it possible to analyze the results of many other sensitizer-wire experiments that in turn point to new directions in our attempts to observe reactive intermediates in the catalytic cycles of P450 and other heme enzymes.
Co-reporter:Jonathan D. Servaites;Sina Yeganeh;Tobin J. Marks
Advanced Functional Materials 2010 Volume 20( Issue 1) pp:97-104
Publication Date(Web):
DOI:10.1002/adfm.200901107
Abstract
Here, means to enhance power conversion efficiency (PCE or η) in bulk-heterojunction (BHJ) organic photovoltaic (OPV) cells by optimizing the series resistance (Rs)—also known as the cell internal resistance—are studied. It is shown that current state-of-the-art BHJ OPVs are approaching the limit for which efficiency can be improved via Rs reduction alone. This evaluation addresses OPVs based on a poly(3-hexylthiophene):6,6-phenyl C61-butyric acid methyl ester (P3HT:PCBM) active layer, as well as future high-efficiency OPVs (η > 10%). A diode-based modeling approach is used to assess changes in Rs. Given that typical published P3HT:PCBM test cells have relatively small areas (∼0.1 cm2), the analysis is extended to consider efficiency losses for larger area cells and shows that the transparent anode conductivity is then the dominant materials parameter affecting Rs efficiency losses. A model is developed that uses cell sizes and anode conductivities to predict current–voltage response as a function of resistive losses. The results show that the losses due to Rs remain minimal until relatively large cell areas (>0.1 cm2) are employed. Finally, Rs effects on a projected high-efficiency OPV scenario are assessed, based on the goal of cell efficiencies >10%. Here, Rs optimization effects remain modest; however, there are now more pronounced losses due to cell size, and it is shown how these losses can be mitigated by using higher conductivity anodes.
Co-reporter:Joseph A. Letizia;Jonathan Rivnay;Antonio Facchetti;Tobin J. Marks
Advanced Functional Materials 2010 Volume 20( Issue 1) pp:50-58
Publication Date(Web):
DOI:10.1002/adfm.200900831
Abstract
The temperature dependence of field-effect transistor (FET) mobility is analyzed for a series of n-channel, p-channel, and ambipolar organic semiconductor-based FETs selected for varied semiconductor structural and device characteristics. The materials (and dominant carrier type) studied are 5,5′′′-bis(perfluorophenacyl)-2,2′:5′,2″:5″,2′′′-quaterthiophene (1, n-channel), 5,5′′′-bis(perfluorohexyl carbonyl)-2,2′:5′,2″:5″,2′′′-quaterthiophene (2, n-channel), pentacene (3, p-channel); 5,5′′′-bis(hexylcarbonyl)-2,2′:5′,2″:5″,2′′′-quaterthiophene (4, ambipolar), 5,5′′′-bis-(phenacyl)-2,2′: 5′,2″:5″,2′′′-quaterthiophene (5, p-channel), 2,7-bis((5-perfluorophenacyl)thiophen-2-yl)-9,10-phenanthrenequinone (6, n-channel), and poly(N-(2-octyldodecyl)-2,2′-bithiophene-3,3′-dicarboximide) (7, n-channel). Fits of the effective field-effect mobility (µeff) data assuming a discrete trap energy within a multiple trapping and release (MTR) model reveal low activation energies (EAs) for high-mobility semiconductors 1–3 of 21, 22, and 30 meV, respectively. Higher EA values of 40–70 meV are exhibited by 4–7-derived FETs having lower mobilities (µeff). Analysis of these data reveals little correlation between the conduction state energy level and EA, while there is an inverse relationship between EA and µeff. The first variable-temperature study of an ambipolar organic FET reveals that although n-channel behavior exhibits EA = 27 meV, the p-channel regime exhibits significantly more trapping with EA = 250 meV. Interestingly, calculated free carrier mobilities (µ0) are in the range of ∼0.2–0.8 cm2 V−1 s−1 in this materials set, largely independent of µeff. This indicates that in the absence of charge traps, the inherent magnitude of carrier mobility is comparable for each of these materials. Finally, the effect of temperature on threshold voltage (VT) reveals two distinct trapping regimes, with the change in trapped charge exhibiting a striking correlation with room temperature µeff. The observation that EA is independent of conduction state energy, and that changes in trapped charge with temperature correlate with room temperature µeff, support the applicability of trap-limited mobility models such as a MTR mechanism to this materials set.
Co-reporter:Neng Guo, Sara A. DiBenedetto, Pratyush Tewari, Michael T. Lanagan, Mark A. Ratner and Tobin J. Marks
Chemistry of Materials 2010 Volume 22(Issue 4) pp:1567
Publication Date(Web):January 5, 2010
DOI:10.1021/cm902852h
A series of 0−3 metal oxide−polyolefin nanocomposites are synthesized via in situ olefin polymerization, using the following single-site metallocene catalysts: C2-symmetric dichloro[rac-ethylenebisindenyl]zirconium(IV), Me2Si(tBuN)(η5-C5Me4)TiCl2, and (η5-C5Me5)TiCl3 immobilized on methylaluminoxane (MAO)-treated BaTiO3, ZrO2, 3-mol %-yttria-stabilized zirconia, 8-mol %-yttria-stabilized zirconia, sphere-shaped TiO2 nanoparticles, and rod-shaped TiO2 nanoparticles. The resulting composite materials are structurally characterized via X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), 13C nuclear magnetic resonance (NMR) spectroscopy, and differential scanning calorimetry (DSC). TEM analysis shows that the nanoparticles are well-dispersed in the polymer matrix, with each individual nanoparticle surrounded by polymer. Electrical measurements reveal that most of these nanocomposites have leakage current densities of ∼10−6−10−8 A/cm2; relative permittivities increase as the nanoparticle volume fraction increases, with measured values as high as 6.1. At the same volume fraction, rod-shaped TiO2 nanoparticle−isotactic polypropylene nanocomposites exhibit significantly greater permittivities than the corresponding sphere-shaped TiO2 nanoparticle−isotactic polypropylene nanocomposites. Effective medium theories fail to give a quantitative description of the capacitance behavior, but do aid substantially in interpreting the trends qualitatively. The energy storage densities of these nanocomposites are estimated to be as high as 9.4 J/cm3.
Co-reporter:JosephA. Letizia Dr.;Scott Cronin;RocioPonce Ortiz Dr.;Antonio Facchetti Dr.;MarkA. Ratner ;TobinJ. Marks
Chemistry - A European Journal 2010 Volume 16( Issue 6) pp:1911-1928
Publication Date(Web):
DOI:10.1002/chem.200901513
Abstract
Electron-transporting organic semiconductors (n-channel) for field-effect transistors (FETs) that are processable in common organic solvents or exhibit air-stable operation are rare. This investigation addresses both these challenges through rational molecular design and computational predictions of n-channel FET air-stability. A series of seven phenacyl–thiophene-based materials are reported incorporating systematic variations in molecular structure and reduction potential. These compounds are as follows: 5,5′′′-bis(perfluorophenylcarbonyl)-2,2′:5′,- 2′′:5′′,2′′′-quaterthiophene (1), 5,5′′′-bis(phenacyl)-2,2′:5′,2′′: 5′′,2′′′-quaterthiophene (2), poly[5,5′′′-(perfluorophenac-2-yl)-4′,4′′-dioctyl-2,2′:5′,2′′:5′′,2′′′-quaterthiophene) (3), 5,5′′′-bis(perfluorophenacyl)-4,4′′′-dioctyl-2,2′:5′,2′′:5′′,2′′′-quaterthiophene (4), 2,7-bis((5-perfluorophenacyl)thiophen-2-yl)-9,10-phenanthrenequinone (5), 2,7-bis[(5-phenacyl)thiophen-2-yl]-9,10-phenanthrenequinone (6), and 2,7-bis(thiophen-2-yl)-9,10-phenanthrenequinone, (7). Optical and electrochemical data reveal that phenacyl functionalization significantly depresses the LUMO energies, and introduction of the quinone fragment results in even greater LUMO stabilization. FET measurements reveal that the films of materials 1, 3, 5, and 6 exhibit n-channel activity. Notably, oligomer 1 exhibits one of the highest μe (up to ≈0.3 cm2 V−1 s−1) values reported to date for a solution-cast organic semiconductor; one of the first n-channel polymers, 3, exhibits μe≈10−6 cm2 V−1 s−1 in spin-cast films (μe=0.02 cm2 V−1 s−1 for drop-cast 1:3 blend films); and rare air-stable n-channel material 5 exhibits n-channel FET operation with μe=0.015 cm2 V−1 s−1, while maintaining a large Ion:off=106 for a period greater than one year in air. The crystal structures of 1 and 2 reveal close herringbone interplanar π-stacking distances (3.50 and 3.43 Å, respectively), whereas the structure of the model quinone compound, 7, exhibits 3.48 Å cofacial π-stacking in a slipped, donor-acceptor motif.
Co-reporter:Xiaodong Chen, Sina Yeganeh, Lidong Qin, Shuzhou Li, Can Xue, Adam B. Braunschweig, George C. Schatz, Mark A. Ratner and Chad A. Mirkin
Nano Letters 2009 Volume 9(Issue 12) pp:3974-3979
Publication Date(Web):November 12, 2009
DOI:10.1021/nl9018726
We report a simple and reproducible method for fabricating heterometallic nanogaps, which are made of two different metal nanorods separated by a nanometer-sized gap. The method is based upon on-wire lithography, which is a chemically enabled technique used to synthesize a wide variety of nanowire-based structures (e.g., nanogaps and disk arrays). This method can be used to fabricate pairs of metallic electrodes, which exhibit distinct work functions and are separated by gaps as small as 2 nm. Furthermore, we demonstrate that a symmetric thiol-terminated molecule can be assembled into such heterometallic nanogaps to form molecular transport junctions (MTJs) that exhibit molecular diode behavior. Theoretical calculations demonstrate that the coupling strength between gold and sulfur (Au−S) is 2.5 times stronger than that of Pt−S. In addition, the structures form Raman hot spots in the gap, allowing the spectroscopic characterization of the molecules that make up the MTJs.
Co-reporter:Sina Yeganeh, Mark A. Ratner, Michael Galperin and Abraham Nitzan
Nano Letters 2009 Volume 9(Issue 5) pp:1770-1774
Publication Date(Web):March 26, 2009
DOI:10.1021/nl803635t
We implement a method to study transport in a basis of many-body molecular states using the nonequilibrium Hubbard Green’s function technique. A well-studied system, a junction consisting of benzene-dithiol on gold, is the focus of our consideration. Electronic structure calculations are carried out at the Hartree−Fock (HF), density functional theory (DFT), and coupled-cluster singles and doubles (CCSD) levels, and multiple molecular states are included in the transport calculation. The conductance calculation yields new information about the transport mechanism in BDT junctions.
Co-reporter:Sara A. DiBenedetto ; Antonio Facchetti ; Mark A. Ratner ;Tobin J. Marks
Journal of the American Chemical Society 2009 Volume 131(Issue 20) pp:7158-7168
Publication Date(Web):May 1, 2009
DOI:10.1021/ja9013166
Developing alternative high dielectric constant (k) materials for use as gate dielectrics is essential for continued advances in conventional inorganic CMOS and organic thin film transistors (OTFTs). Thicker films of high-k materials suppress tunneling leakage currents while providing effective capacitances comparable to those of thin films of lower-k materials. Self-assembled monolayers (SAMs) and multilayers offer attractive options for alternative OTFT gate dielectrics. One class of materials, organosilane-based self-assembled nanodielectrics (SANDs), has been shown to form robust films with excellent insulating and surface passivation properties, enhancing both organic and inorganic TFT performance and lowering device operating voltages. Since gate leakage current through the dielectric is one factor limiting continued TFT performance improvements, we investigate here the current (voltage, temperature) (I (V,T)) transport characteristics of SAND types II (π-conjugated layer) and III (σ-saturated + π-conjugated layers) in Si/native SiO2/SAND/Au metal−insulator−metal (MIS) devices over the temperature range −60 to +100 °C. It is found that the location of the π-conjugated layer with respect to the Si/SiO2 substrate surface in combination with a saturated alkylsilane tunneling barrier is crucial in controlling the overall leakage current through the various SAND structures. For small applied voltages, hopping transport dominates at all temperatures for the π-conjugated system (type II). However, for type III SANDs, the σ- and π- monolayers dominate the transport in two different transport regimes: hopping between +25 °C and +100 °C, and an apparent switch to tunneling for temperatures below 25 °C. The σ-saturated alkylsilane tunneling barrier functions to reduce type III current leakage by blocking injected electrons, and by enabling bulk-dominated (Poole−Frenkel) transport vs electrode-dominated (Schottky) transport in type II SANDs. These observations provide insights for designing next-generation self-assembled gate dielectrics, since the bulk-dominated transport resulting from combining σ- and π-layers should enable realization of gate dielectrics with further enhanced performance.
Co-reporter:Jun Liu, Alexander W. Hains, Jonathan D. Servaites, Mark A. Ratner and Tobin J. Marks
Chemistry of Materials 2009 Volume 21(Issue 21) pp:5258
Publication Date(Web):October 12, 2009
DOI:10.1021/cm902265n
Highly conductive In-doped CdO/Sn-doped In2O3 (CIO/ITO) bilayer transparent conducting oxide (TCO) thin films were prepared by combining, in sequence, metal-organic chemical vapor deposition (MOCVD) and ion-assisted deposition (IAD) techniques. The bilayer substrates, with a low In content of ∼19 atom % and a low sheet resistance of only ∼4.9 Ω/◻, were investigated as anodes in the bulk-heterojunction (BHJ) organic photovoltaic (OPV) devices using poly(2-methoxy-5-(3′,7′-dimethyloctyloxy)-1,4-phenylenevinylene) (MDMO-PPV):[6,6]-phenyl C61 butyric acid methyl ester (PCBM) as the active layer. The bilayer anode OPVs of the current laboratory size (∼0.06 cm2) exhibit performance comparable to those of commercial ITO-based control devices. The effect of TCO conductivity on OPV performance in larger area devices is analyzed through a simulation model. The results reveal significant advantages of using the highly conductive bilayer TCO anodes for large-area OPV cells.
Co-reporter:Xiaodong Chen Dr.;AdamB. Braunschweig Dr.;MichaelJ. Wiester;Sina Yeganeh;MarkA. Ratner ;ChadA. Mirkin
Angewandte Chemie International Edition 2009 Volume 48( Issue 28) pp:5178-5181
Publication Date(Web):
DOI:10.1002/anie.200806028
Co-reporter:Xiaodong Chen Dr.;AdamB. Braunschweig Dr.;MichaelJ. Wiester;Sina Yeganeh;MarkA. Ratner ;ChadA. Mirkin
Angewandte Chemie 2009 Volume 121( Issue 28) pp:
Publication Date(Web):
DOI:10.1002/ange.200990146
Co-reporter:Xiaodong Chen Dr.;AdamB. Braunschweig Dr.;MichaelJ. Wiester;Sina Yeganeh;MarkA. Ratner ;ChadA. Mirkin
Angewandte Chemie 2009 Volume 121( Issue 28) pp:5280-5283
Publication Date(Web):
DOI:10.1002/ange.200806028
Co-reporter:Xiaodong Chen Dr.;AdamB. Braunschweig Dr.;MichaelJ. Wiester;Sina Yeganeh;MarkA. Ratner ;ChadA. Mirkin
Angewandte Chemie International Edition 2009 Volume 48( Issue 28) pp:
Publication Date(Web):
DOI:10.1002/anie.200990144
Co-reporter:Matthew G. Reuter, Thorsten Hansen, Tamar Seideman and Mark A. Ratner
The Journal of Physical Chemistry A 2009 Volume 113(Issue 16) pp:4665-4676
Publication Date(Web):March 26, 2009
DOI:10.1021/jp811492u
Analytical self-energies for molecular interfaces with one-dimensional, tight-binding semiconductors are derived, along with analytical solutions to the electrode eigensystems. These models capture the fundamental differences between the transport properties of metals and semiconductors and also account for the appearance of surface states. When the models are applied to zero-temperature electrode−molecule−electrode conductance, junctions with two semiconductor electrodes exhibit a minimum bias threshold for generating current due to the absence of electrode states near the Fermi level. Molecular interactions with semiconductor electrodes additionally produce (i) non-negligible molecular-level shifting by mechanisms absent in metals and (ii) sensitivity of the transport to the semiconductor−molecule bonding configuration. Finally, the general effects of surface states on molecular transport are discussed.
Co-reporter:Joseph A. Letizia ; Michael R. Salata ; Caitlin M. Tribout ; Antonio Facchetti ; Mark A. Ratner ;Tobin J. Marks
Journal of the American Chemical Society 2008 Volume 130(Issue 30) pp:9679-9694
Publication Date(Web):July 2, 2008
DOI:10.1021/ja710815a
Electron transporting (n-channel) polymer semiconductors for field-effect transistors are rare. In this investigation, the synthesis and characterization of new electron-depleted N-alkyl-2,2′-bithiophene-3,3′-dicarboximide-based π-conjugated homopolymers and copolymers containing the 2,2′-bithiophene unit are reported. A novel design approach is employed using computational modeling to identify favorable monomer properties such as core planarity, solubilizing substituent tailorability, and appropriate electron affinity with gratifying results. Monomeric model compounds are synthesized to confirm these properties, and a crystal structure reveals a short 3.43 Å π−π stacking distance with favorable solubilizing substituent orientations. A family of 10 homopolymers and bithiophene copolymers is then synthesized via Yamamoto and Stille polymerizations, respectively. Two of these polymers are processable in common organic solvents: the homopolymer poly(N-(2-octyldodecyl)-2,2′-bithiophene-3,3′-dicarboximide) (P1) exhibits n-channel FET activity, and the copolymer poly(N-(2-octyldodecyl)-2,2′:5′,2′′:5′′,2′′′-quaterthiophene-3,3′-dicarboximide) (P2) exhibits air-stable p-channel FET operation. After annealing, P1 films exhibit a very high degree of crystallinity and an electron mobility > 0.01 cm2 V−1 s−1 with a current on−off ratio of 107, which is remarkably independent of film-deposition conditions. Extraordinarily, P1 films also exhibit terracing in AFM images with a step height matching the X-ray diffraction d spacing, a rare phenomenon for polymeric organic semiconductors. Another fascinating property of these materials is the air-stable p-channel FET performance of annealed P2 films, which exhibit a hole mobility of ∼0.01 cm2 V−1 s−1 and a current on−off ratio of 107.
Co-reporter:Randall H. Goldsmith, Orlando DeLeon, Thea M. Wilson, Daniel Finkelstein-Shapiro, Mark A. Ratner and Michael R. Wasielewski
The Journal of Physical Chemistry A 2008 Volume 112(Issue 19) pp:4410-4414
Publication Date(Web):April 18, 2008
DOI:10.1021/jp801084v
The temperature dependence of intramolecular charge separation in a series of donor-bridge-acceptor molecules having phenothiazine (PTZ) donors, 2,7-oligofluorene FLn (n = 1−4) bridges, and perylene-3,4:9,10-bis(dicarboximide) (PDI) acceptors was studied. Photoexcitation of PDI to its lowest excited singlet state results in oxidation of PTZ via the FLn bridge. In toluene, the temperature dependence of the charge separation rate constants for PTZ-FLn-PDI, (n = 1−4) is relatively weak and is successfully described by the semiclassical Marcus equation. The activation energies for charge separation suggest that bridge charge carrier injection is not the rate limiting step. The difficulty of using temperature and length dependence to differentiate hopping and superexchange is discussed, with difficulties in the latter topic explored via an extension of a kinetic model proposed by Bixon and Jortner.
Co-reporter:Brett M. Savoie ; Akshay Rao ; Artem A. Bakulin ; Simon Gelinas ; Bijan Movaghar ; Richard H. Friend ; Tobin J. Marks
Journal of the American Chemical Society () pp:
Publication Date(Web):January 24, 2014
DOI:10.1021/ja411859m
Natural photosynthetic complexes accomplish the rapid conversion of photoexcitations into spatially separated electrons and holes through precise hierarchical ordering of chromophores and redox centers. In contrast, organic photovoltaic (OPV) cells are poorly ordered, utilize only two different chemical potentials, and the same materials that absorb light must also transport charge; yet, some OPV blends achieve near-perfect quantum efficiency. Here we perform electronic structure calculations on large clusters of functionalized fullerenes of different size and ordering, predicting several features of the charge generation process, outside the framework of conventional theories but clearly observed in ultrafast electro-optical experiments described herein. We show that it is the resonant coupling of photogenerated singlet excitons to a high-energy manifold of fullerene electronic states that enables efficient charge generation, bypassing localized charge-transfer states. In contrast to conventional views, our findings suggest that fullerene cluster size, concentration, and dimensionality control charge generation efficiency, independent of exciton delocalization.
Co-reporter:Brett M. Savoie, Nicholas E. Jackson, Tobin J. Marks and Mark A. Ratner
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 13) pp:NaN4547-4547
Publication Date(Web):2013/02/01
DOI:10.1039/C3CP50438G
We present results showing that common approximations employed in the design and characterization of organic photovoltaic (OPV) materials can lead to significant errors in widely adopted design rules. First, we assess the validity of the common practice of using HOMO and LUMO energies in place of formal redox potentials to characterize organic semiconductors. We trace the formal justification for this practice and survey its limits in a way that should be useful for those entering the field. We find that while the HOMO and LUMO energies represent useful descriptive approximations, they are too quantitatively inaccurate for predictive material design. Second, we show that the excitonic nature of common organic semiconductors makes it paramount to distinguish between the optical and electronic bandgaps for materials design. Our analysis shows that the usefulness of the “LUMO–LUMO Offset” as a design parameter for exciton dissociation is directly tied to the accuracy of the one-electron approximation. In particular, our results suggest that the use of the “LUMO–LUMO Offset” as a measure of the driving force for exciton dissociation leads to a systematic overestimation that should be cautiously avoided.
Co-reporter:Lauren M. Matosziuk, Robert J. Holbrook, Lisa M. Manus, Marie C. Heffern, Mark A. Ratner and Thomas J. Meade
Dalton Transactions 2013 - vol. 42(Issue 11) pp:NaN4012-4012
Publication Date(Web):2013/01/22
DOI:10.1039/C2DT32565A
Cobalt(III) Schiff base complexes, such as [Co(acacen)L2]+, inhibit the function of Zn(II)-dependent proteins through dissociative exchange of the axial ligands with key histidine residues of the target protein. Consequently the efficacy of these compounds depends strongly on the lability of the axial ligands. A series of [Co(acacen)L2]+ complexes with various axial ligands was investigated using DFT to determine the kinetics and thermodynamics of ligand exchange and hydrolysis. Results showed excellent agreement with experimental data, indicating that axial ligand lability is determined by several factors: pKa of the axial ligand, the kinetic barrier to ligand dissociation, and the relative thermodynamic stability of the complexes before and after exchange. Hammett plots were constructed to determine if the kinetics and thermodynamics of exchange can be modulated by the addition of an electron-withdrawing group (EWG) to either the axial ligand itself or to the equatorial acacen ligand. Results predict that addition of an EWG to the axial ligand will shift the kinetics and thermodynamics so as to promote axial ligand exchange, while addition of an EWG to acacen will decrease axial ligand lability. These investigations will aid in the design of the next generation of [Co(acacen)L2]2+, allowing researchers to develop new, more effective inhibitors.
Co-reporter:Guangqi Li, Abraham Nitzan and Mark A. Ratner
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 41) pp:NaN14276-14276
Publication Date(Web):2012/08/10
DOI:10.1039/C2CP41532A
A simple model is constructed to describe dissociation of charge transfer excitons in bulk heterojunction solar cells, and its dependence on the physical parameters of the system. In bulk heterojunction organic photovoltaics (OPVs), exciton dissociation occurs almost exclusively at the interface between the donor and acceptor, following one-electron initial excitation from the HOMO to the LUMO levels of the donor, and charge transfer to the acceptor to make a charge-transfer exciton. After exciton breakup, and neglecting the trapping of individual carriers, the electron may undergo two processes for decay: one process involves the electron and/or hole leaving the interface, and migrating to the electrode. This is treated here as the electron moving on a set of acceptor sites. The second loss process is radiationless decay following recombination of the acceptor electron with the donor cation; this is treated by adding a relaxation term. These two processes compete with one another. We model both the exciton breakup and the subsequent electron motion. Results depend on tunneling amplitude, energetics, disorder, Coulomb barriers, and energy level matchups, particularly the so-called LUMO–LUMO offset.
.jpg)