Co-reporter:Ekram Hossain, Shihu M. Deng, Samer Gozem, Anna I. Krylov, Xue-Bin Wang, and Paul G. Wenthold
Journal of the American Chemical Society August 16, 2017 Volume 139(Issue 32) pp:11138-11138
Publication Date(Web):July 21, 2017
DOI:10.1021/jacs.7b05197
Structures and energetics of o-, m-, and p-quinonimide anions (OC6H4N–) and quinoniminyl radicals have been investigated by using negative ion photoelectron spectroscopy. Modeling of the photoelectron spectrum of the ortho isomer shows that the ground state of the anion is a triplet, while the quinoniminyl radical has a doublet ground state with a doublet-quartet splitting of 35.5 kcal/mol. The para radical has doublet ground state, but a band for a quartet state is missing from the photoelectron spectrum indicating that the anion has a singlet ground state, in contrast to previously reported calculations. The theoretical modeling is revisited here, and it is shown that accurate predictions for the electronic structure of the para-quinonimide anion require both an accurate account of electron correlation and a sufficiently diffuse basis set. Electron affinities of o- and p-quinoniminyl radicals are measured to be 1.715 ± 0.010 and 1.675 ± 0.010 eV, respectively. The photoelectron spectrum of the m-quinonimide anion shows that the ion undergoes several different rearrangements, including a rearrangement to the energetically favorable para isomer. Such rearrangements preclude a meaningful analysis of the experimental spectrum.
Co-reporter:Nathan J. Rau, Paul G. Wenthold
International Journal of Mass Spectrometry 2015 Volume 377() pp:496-501
Publication Date(Web):1 February 2015
DOI:10.1016/j.ijms.2014.05.008
•We have examined the chemistry of deprotonated imines (imides).•Reactions with CS2 proceed by S/N exchange and hydride transfer.•Reactions with NO show that the p-nitro-substituted ion has a low-lying triplet state.•Electronic structure calculations also predict a triplet state for p-nitro.•p-Nitrobenzaldimide is a rare example of a low-energy triplet anion.Ion–molecule reactions show that p-nitrobenzaldimide, the conjugate-base anion of the benzaldimine, has a low-lying triplet state that is accessible under thermal conditions. Whether it is the ground state or a low-lying excited state cannot be determined, and both possibilities have computational support. Reactivity studies indicate that imide anions with weaker π-donors have singlet ground states, with no evidence for an accessible triplet.
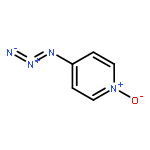
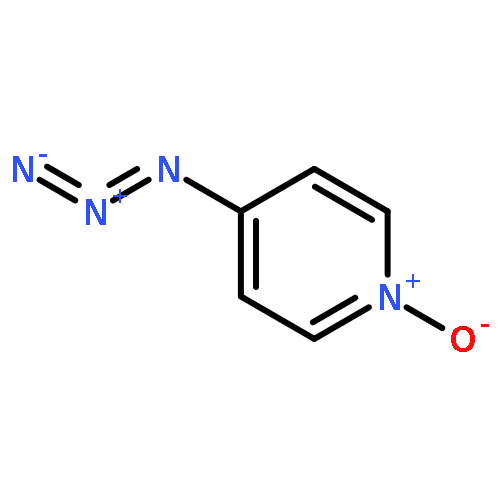
Co-reporter:Damodar Koirala, James S. Poole, Paul G. Wenthold
International Journal of Mass Spectrometry 2015 Volume 378() pp:69-75
Publication Date(Web):15 February 2015
DOI:10.1016/j.ijms.2014.07.017
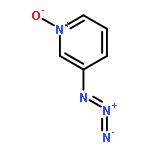
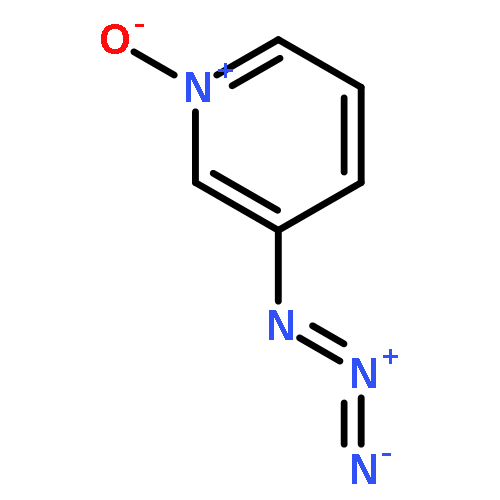
•We have examined the ion–molecule reactions of pyridinylnitrene-n-oxide anions.•CS2 reacts at the nitrene nitrogen in 3-pyridinylnitrene-n-oxide anion.•With 4-pyridinylnitrene-n-oxide anion, CS2 reacts at both the nitrene N and at the O.•Resonance interaction makes the n-oxide more reactive in the 4-isomer.Ion–molecule reactions in a flowing afterglow are used to examine the electronic structure of 3- and 4-pyridinylnitrene-n-oxide radical anions. Reactions with nitric oxide are generally similar to those reported previously for other aromatic nitrene radical anions. In particular, phenoxide formation by nitrogen–oxygen exchange is observed with both isomers. Oxygen atom abstraction by NO is also observed with both isomers. Very significant differences in the reactivity are observed in the reactions of the two isomers with carbon disulfide. The reactivity of the 3-n-oxide isomer with CS2 is similar to that observed previously for nitrene radical anions, and reactions of the n-oxide moiety are not observed, similar to what is expected based on solution chemistry. The 4-n-oxide isomer, however, undergoes many reactions, including oxygen atom and oxygen ion transfer and sulfur–oxygen exchange, that involve the n-oxide oxygen. The increased reactivity of the oxygen is attributed to increased charge density at the oxygen due to pi electron donation of the nitrene anion in the para position.
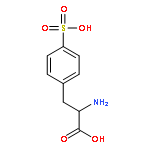
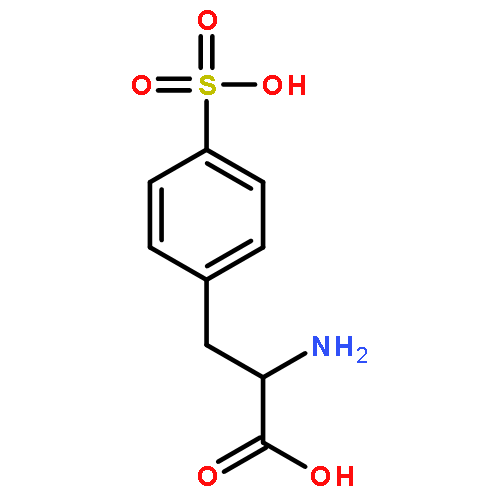
Co-reporter:Damodar Koirala;Sampath Ranasinghe Kodithuwakkuge
Journal of Physical Organic Chemistry 2015 Volume 28( Issue 10) pp:635-644
Publication Date(Web):
DOI:10.1002/poc.3464
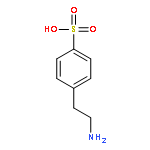
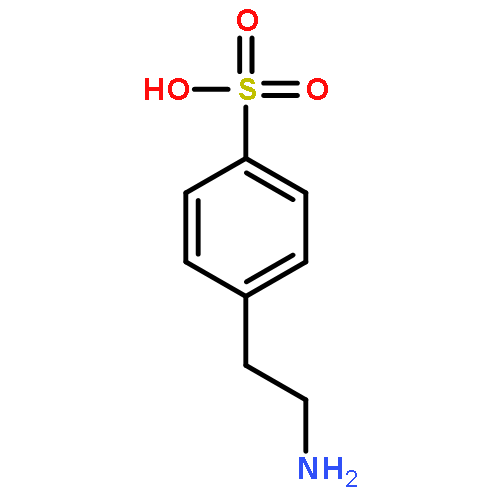
The dissociation pathways of a gas-phase amino acid with a canonical (non-zwitterionic) α-amino acid moiety are studied by using mass spectrometry. Investigation of the canonical amino acid moiety is possible because the ionized amino acid, a sulfonated phenylalanine, has a charge center that is separated from the amino acid, and dissociation occurs by charge-remote fragmentation. The amino acid is found to dissociate only by loss of NH3 upon collision-induced dissociation to form a substituted α-lactone. The dissociation is consistent with what has been observed previously upon pyrolysis of other α-substituted carboxylic acids. Decarboxylation, which has also been reported previously for amino acid pyrolysis, is not observed, likely because the product would be a high-energy, ammonium ylide. The resulting α-lactone is found to undergo dissociation by decarbonylation to give an aldehyde, and by loss of CO2. Decarboxylation is calculated to occur through a transition state involving hydride shift coupled with lactone ring-opening. The transition state is found to be stabilized by the negative charge, and therefore, decarboxylation is more favorable for anions. The results show that remote ionic groups can be used as mostly inert charge carriers to enable mass spectrometry to be used to investigate the gas-phase physical and chemical properties of different types of functional groups, including amino acids. Copyright © 2015 John Wiley & Sons, Ltd.
Co-reporter:Laura J. Haupert and Paul G. Wenthold
The Journal of Physical Chemistry A 2013 Volume 117(Issue 6) pp:1164-1170
Publication Date(Web):July 24, 2012
DOI:10.1021/jp305119y
The hydration energies of aromatic ions, measured by using energy-resolved collision-induced dissociation measurements, are reported. The hydration energies of protonated acetophenone, aniline, anisole, benzene, benzonitrile, phenol, and toluene are 0.67 ± 0.04, 0.62 ± 0.04, 0.86 ± 0.04, 0.46 ± 0.06, 0.84 ± 0.04, 0.75 ± 0.07, and 0.42 ± 0.04 eV, respectively. The measured values are in good agreement with those predicted by using coupled cluster theory, provided the proper geometries of the ions and sufficient basis set were used.
Co-reporter:Nathan J. Rau ; Emily A. Welles
Journal of the American Chemical Society 2012 Volume 135(Issue 2) pp:683-690
Publication Date(Web):December 31, 2012
DOI:10.1021/ja306364z
The electronic structures of phenylnitrenes with anionic π-donating substituents are investigated by using mass spectrometry and electronic structure calculations. Reactions of para-CH2–-substituted phenylnitrene, formed by dissociative deprotonation of p-azidotoluene, with CS2 and NO indicate that it has a closed-shell singlet ground state, whereas reactions of p-oxidophenylnitrene formed by dissociative deprotonation of p-azidophenol indicate either a triplet ground state or a singlet with a small singlet–triplet splitting. The ground electronic state assignments based on ion reactivity are consistent with electronic structure calculations. The stability of the closed-shell singlet states in nitrenes is shown by Natural Resonance Theory to be very sensitive to the amount of deprotonated-imine character in the wave function, such that large changes in state energies can be achieved by small modifications of the electronic structure.
Co-reporter:Paul G. Wenthold
The Journal of Organic Chemistry 2012 Volume 77(Issue 1) pp:208-214
Publication Date(Web):November 7, 2011
DOI:10.1021/jo2016967
Geometries and energies of the triplet and singlet states of 2-furanylnitrene and 3-furanylnitrene have been calculated by using spin–flip coupled-cluster methods. Calculations with triple-ζ basis sets predict a singlet–triplet splitting of 10.9 kcal/mol for 2-furanylnitrene, 4.5 kcal/mol smaller than that in phenylnitrene. In contrast, the singlet–triplet splitting in 3-furanylnitrene is computed to be 1.9 kcal/mol larger than that in phenylnitrene. The differences in the singlet–triplet splittings for the furanylnitrenes are attributed to the differences in the radical stabilizing abilities of the 2-furanyl- and 3-furanyl-groups compared to a phenyl ring. Comparison of the singlet–triplet splittings of more than 20 substituted aromatic nitrenes and the radical stabilizing ability of the aromatic systems reveals a high degree of correlation between the singlet–triplet splitting and the radical stabilizing ability, indicating that singlet states of aromatic nitrenes are preferentially stabilized by radical stabilizing substituents. The preferential stabilization of the singlet states is attributed to the decrease in electron pair repulsion resulting from increased delocalization of the radical electron.
Co-reporter:Jamelle K.P. Williams, Paul G. Wenthold
International Journal of Mass Spectrometry 2011 Volume 299(Issue 1) pp:9-12
Publication Date(Web):1 January 2011
DOI:10.1016/j.ijms.2010.09.003
The fluoride affinities of fluorinated alanes, AlHmF3−m (m = 1–3) were measured using energy-resolved collision-induced dissociation of fluorinated aluminate anions. The AlHmF4−m− anions were formed by reaction of dimethylethylamine-alane with fluoride ion and F2. From the measured bond dissociation energies, the fluoride affinities of fluorinated alanes are determined to be 93.2 ± 3.1, 97.5 ± 4.0, and 108.6 ± 3.7 kcal/mol for m = 3, 2, and 1, respectively. The fluoride affinities are in good agreement with the theoretical calculations at the CCSD(T)/CBS and B3LYP/6-31 + G* levels of theory. The increased Lewis acidity of more fluorinated alanes is attributed to increased positive charge density on the aluminum.Graphical abstractResearch highlights▶ Fluorinated aluminate anions (AlHmF4−m−) can be formed in the gas phase by ionization of diethylmethylamine-alane with molecular fluorine. ▶ Energy-resolved collision-induced dissociation is used to measure fluoride affinities of the alanes. ▶ The measured fluoride affinities agree with predictions obtained from coupled-cluster calculations with very large basis sets. ▶ Fluoride affinities reflect the extent of positive charge character on the aluminum in the alanes.
Co-reporter:Nathan J. Rau and Paul G. Wenthold
The Journal of Physical Chemistry A 2011 Volume 115(Issue 37) pp:10353-10362
Publication Date(Web):August 3, 2011
DOI:10.1021/jp2051068
The absolute enthalpies of formation of 3,4-, 2,3-, and/or 2,4-didehydropyridines (3,4-, 2,3- and 2,4-pyridynes) have been determined by using energy-resolved collision-induced dissociation of deprotonated 2- and 3-chloropyridines. Bracketing experiments find the gas-phase acidities of 2- and 3-chloropyridines to be 383 ± 2 and 378 ± 2 kcal/mol, respectively. Whereas deprotonation of 3-chloropyridine leads to formation of a single ion isomer, deprotonation of the 2-chloro isomer results in a nearly 60:40 mixture of regioisomers. The enthalpy of formation of 3,4-pyridyne is measured to be 121 ± 3 kcal/mol by using the chloride dissociation energy for deprotonated 3-chloropyridine. The structure of the product formed upon dissociation of the ion from 2-chloropyridine cannot be unequivocally assigned because of the isomeric mixture of reactant ions and the fact that the potential neutral products (2,3-pyridyne and 2,4-pyridyne) are predicted by high level spin-flip coupled-cluster calculations to be nearly the same in energy. Consequently, the enthalpies of formation for both neutral products are assigned to be 130 ± 3 kcal/mol. Comparison of the enthalpies of dehydrogenation of benzene and pyridine indicates that the nitrogen in the pyridine ring does not have any effect on the stability of the aryne triple bond in 3,4-pyridyne, destabilizes the aryne triple bond in 2,3-pyridyne, and stabilizes the 1,3-interaction in 2,4-pyridyne compared to that in m-benzyne. Natural bond order calculations show that the effects on the 2,3- and 2,4-pyridynes result from polarization of the electrons caused by interaction with the lone pair. The polarization in 2,4-pyridyne is stabilizing because it creates a 1,2-interaction between the nitrogen and dehydrocarbons that is stronger than the 1,3-interaction between the dehydrocarbons.
Co-reporter:Matthew J. Lenington and Paul G. Wenthold
The Journal of Physical Chemistry A 2010 Volume 114(Issue 3) pp:1334-1337
Publication Date(Web):September 10, 2009
DOI:10.1021/jp905757p
The meta- and para-bis-allylbenzene radical anions have been generated and investigated using mass spectrometry. The ions are formed by reaction of the corresponding bis-2-propenylbenzenes with atomic oxygen anion. Reactivity of the ions indicates that the ions most likely have a bis-allylbenzene structure. Reaction of the ions with carbon disulfide creates CS2 adducts, which, upon collision-induced dissociation, decompose to regenerate the bis-allylbenzene anion or carbon disulfide radical anion. The branching ratios for the two products indicate differences in the electronic structures of the neutral bis-allylbenzene diradicals. The difference in branching ratios and corresponding estimated electron affinities is interpreted in terms of different electronic states being formed, with the para diradical a singlet and the meta diradical either a ground-state triplet or a singlet with a very small singlet−triplet splitting. The difference in electron affinities is used to estimate a singlet−triplet splitting of 0.06 eV for the para diradical. The studies show that topology can be used to control the electronic properties of disjoint, tetramethyleneethane (TME)-like diradicals.
Co-reporter:Neloni R. Wijeratne
Journal of The American Society for Mass Spectrometry 2007 Volume 18( Issue 11) pp:2014-2016
Publication Date(Web):2007 November
DOI:10.1016/j.jasms.2007.08.017
Benzoylnitrene radical anion, formed in high abundance by electron ionization of benzoylazide, is found to be a useful reagent for the formation of ionized reactive intermediates, such as diradicals and carbenes. The reactivity of the ion is similar to what is observed with atomic oxygen anion, occurring in many instances by H2+ transfer. However, because benzoylnitrene radical anion is less basic than oxygen anion, it undergoes H2+ transfer with substrates that react with oxygen anion by only proton transfer and therefore can be used to generate reactive ions not easily prepared by other methodologies.
Co-reporter:Silvi A. Chacko, Ian H. Krouse, Loubna A. Hammad, Paul G. Wenthold
Journal of the American Society for Mass Spectrometry 2006 Volume 17(Issue 1) pp:51-55
Publication Date(Web):January 2006
DOI:10.1016/j.jasms.2005.08.018
Hydrogen cyanide (HCN) for use in ion preparation can be generated in the gas phase by the neutral–neutral reaction of trimethylsilyl cyanide (Me3SiCN) and water in a flowing afterglow mass spectrometer. We demonstrate that the approach can be used to generate a wide range of HCN solvated ions such as F−(HCN), Cl−(HCN), CN−(HCN), PhNO2·−(HCN), Me3SiO−(HCN),and PhSiF4−(HCN), many of which are otherwise difficult to generate. The bond dissociation energy of CN−(HCN), generated by using this approach, has been measured by using energy-resolved collision-induced issociation (CID) to be 0.87 ± 0.07 eV.
Co-reporter:Ian H. Krouse, Paul G. Wenthold
Journal of the American Society for Mass Spectrometry 2005 Volume 16(Issue 5) pp:697-707
Publication Date(Web):May 2005
DOI:10.1016/j.jasms.2005.01.014
In this study, preparation and decomposition of five novel pentavalent fluorosiliconates, RSi(CH3)3F− (R = CH3CH2O, CF3CH2O, (CH3)2CHO, (CH3)3SiO, and (CH3)3SiNH) is used to investigate the process of fluoride-induced desilylation. The siliconates were characterized by collision-induced dissociation and energy-resolved mass spectrometry. Decomposition of RSi(CH3)3F− leads to loss of the nucleophile R− and FSi(CH3)3, except in the case of (CH3)3SiNHSi(CH3)3F−, where HF loss is also observed. Ion affinities for FSi(CH3)3 have been measured for all five nucleophiles, and compare well with computational predictions. The observed trend of the bond dissociation energies resembles the trend of ΔHacid values for the corresponding conjugate acids, RH. Additionally, this data has been incorporated with existing thermochemistry to derive fluoride affinities for four of the silanes (R = CH3CH2O, (CH3)2CHO, (CH3)3SiO, and (CH3)3SiNH). We use the fluoride affinity of the silanes and the FSi(CH3)3 affinity of the departing nucleophilic anion to assess the feasibility of fluoride-induced desilylation of the silanes examined in this work.
Co-reporter:Paul G. Wenthold
Angewandte Chemie 2005 Volume 117(Issue 44) pp:
Publication Date(Web):8 NOV 2005
DOI:10.1002/ange.200502644
Mehrfacher Bindungsbruch: Der kürzlich erbrachte Nachweis und die Charakterisierung des 1,2,3-Tridehydrobenzoltriradikals (siehe Schema) sind ein weiterer Schritt hin zur Aufschlüsselung des systematischen Benzolabbaus. Die Ergebnisse geben Auskunft über die elektronische Struktur des Triradikals und werden mit bisherigen experimentellen und theoretischen Studien abgeglichen.
Co-reporter:Paul G. Wenthold
Angewandte Chemie International Edition 2005 44(44) pp:7170-7172
Publication Date(Web):
DOI:10.1002/anie.200502644
Co-reporter:Lyudmila V. Slipchenko;Tamara E. Munsch ;Anna I. Krylov
Angewandte Chemie 2004 Volume 116(Issue 6) pp:
Publication Date(Web):27 JAN 2004
DOI:10.1002/ange.200490009
Co-reporter:Lyudmila V. Slipchenko;Tamara E. Munsch ;Anna I. Krylov
Angewandte Chemie International Edition 2004 Volume 43(Issue 6) pp:
Publication Date(Web):27 JAN 2004
DOI:10.1002/anie.200490009
Co-reporter:Lyudmila V. Slipchenko;Tamara E. Munsch ;Anna I. Krylov
Angewandte Chemie International Edition 2004 Volume 43(Issue 6) pp:
Publication Date(Web):8 JAN 2004
DOI:10.1002/anie.200352990
Theory predicts an open-shell doublet ground state for the 5-dehydro-1,3-quinodimethane triradical (see picture), with three low-spin coupled electrons in three singly occupied molecular orbitals. The bond dissociation enthalpy for formation of the triradical from meta-xylylene, measured by MS indicates an interaction of (1±4) kcal mol−1 between the unpaired electrons in the σ and π systems in the triradical.
Co-reporter:Lyudmila V. Slipchenko;Tamara E. Munsch ;Anna I. Krylov
Angewandte Chemie 2004 Volume 116(Issue 6) pp:
Publication Date(Web):8 JAN 2004
DOI:10.1002/ange.200352990
Gemäß theoretischer Vorhersagen hat das 5-Dehydro-1,3-chinodimethan-Triradikal (siehe Bild) einen offenschaligen Grundzustand, in dem die drei Elektronen in einfach besetzten MOs zu einer Low-Spin-Konfiguration gekoppelt sind. Die massenspektrometrische Bestimmung der Bindungsdissozationsenthalpie für die Bildung des Triradikals aus meta-Xylol ergab eine Wechselwirkungsenergie von (1±4) kcal mol−1 zwischen den ungepaarten Elektronen des σ- und des π-Systems.
Co-reporter:Haiqing Hu, Paul G. Wenthold
Journal of the American Society for Mass Spectrometry 2001 Volume 12(Issue 7) pp:840-845
Publication Date(Web):July 2001
DOI:10.1016/S1044-0305(01)00252-5
The structure of ionized 1,5-hexadiene, prepared by charge transfer between 1,5-hexadiene and CS2+·, is examined using energy-resolved collision-induced dissociation (CID). By comparing the product distributions and product appearance curves with those of authentic low-energy C6H10+· ions, it is determined that 1,5-hexadiene cation spontaneously rearranges to cyclohexene cation in the gas-phase. The proposed mechanism for formation of cyclohexene cation in the gas phase is analogous to that determined for this process under matrix isolation conditions, where it proceeds via a Cope rearrangement to the cyclohexane-1,4-diyl cation, followed by isomerization to cyclohexene cation. It is shown that electron ionization (EI) of 1,5-hexadiene gives a different molecular ion than is obtained upon chemical ionization (CI). The energy-resolved CID mass spectrum for the EI product is consistent with what would be obtained for a mixture of low energy ion isomers.
Co-reporter:Paul G. Wenthold, Xinping Liu
International Journal of Mass Spectrometry 2001 Volume 207(1–2) pp:69-72
Publication Date(Web):12 April 2001
DOI:10.1016/S1387-3806(01)00356-6
Isotopic labeling and energy-resolved collision-induced dissociation (CID) experiments show that methyl cation loss from protonated acetaldehyde occurs by at least two different mechanisms, one involving direct cleavage and the second involving rearrangement. A statistical scrambling mechanism is ruled out from the CID appearance curves for the CH3+ and CH2D+ products from CH3C(OD)H+, whereas a possible kinetic isotope effect is ruled out by using the alternately labeled CD3C(OH)H+ acetaldehyde ion. The results show that dissociation of protonated aldehydes is even more complex than is suggested by standard CID spectra.
Co-reporter:Paul G. Wenthold
Journal of the American Society for Mass Spectrometry 2000 Volume 11(Issue 7) pp:601-605
Publication Date(Web):July 2000
DOI:10.1016/S1044-0305(00)00120-3
The proton affinities bromo- and iodoacetonitrile are determined using a full implementation of the kinetic method, which includes calculation of the entropies. Branching ratios for dissociation of proton-bound dimers are measured for collision energies ranging from ∼2 to 5 eV. Using a rigorously correct statistical approach, the proton affinities of bromo- and iodoacetonitrile are calculated to be 179.8 ± 1.7 and 182.9 ± 1.6 kcal/mol, respectively. It is shown that neglecting the entropy contributions for these systems leads to proton affinities that are too high by ∼0.6 kcal/mol.
Co-reporter:Neloni R. Wijeratne, Paul G. Wenthold
Journal of the American Society for Mass Spectrometry (November 2007) Volume 18(Issue 11) pp:2014-2016
Publication Date(Web):1 November 2007
DOI:10.1016/j.jasms.2007.08.017
Benzoylnitrene radical anion, formed in high abundance by electron ionization of benzoylazide, is found to be a useful reagent for the formation of ionized reactive intermediates, such as diradicals and carbenes. The reactivity of the ion is similar to what is observed with atomic oxygen anion, occurring in many instances by H2+ transfer. However, because benzoylnitrene radical anion is less basic than oxygen anion, it undergoes H2+ transfer with substrates that react with oxygen anion by only proton transfer and therefore can be used to generate reactive ions not easily prepared by other methodologies.

![Benzonitrile, 4-[[(trimethylsilyl)imino]methyl]-](http://img.cochemist.com/ccimg/199700/199604-07-4.png)
![Benzonitrile, 4-[[(trimethylsilyl)imino]methyl]-](http://img.cochemist.com/ccimg/199700/199604-07-4_b.png)
![Silanamine, 1,1,1-trimethyl-N-[(4-nitrophenyl)methylene]-](http://img.cochemist.com/ccimg/160400/160313-81-5.png)
![Silanamine, 1,1,1-trimethyl-N-[(4-nitrophenyl)methylene]-](http://img.cochemist.com/ccimg/160400/160313-81-5_b.png)
![2-Propenoic acid, 3-[4-(chlorosulfonyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/155400/155378-73-7.png)
![2-Propenoic acid, 3-[4-(chlorosulfonyl)phenyl]-, (2E)-](http://img.cochemist.com/ccimg/155400/155378-73-7_b.png)
![Silanamine, 1,1,1-trimethyl-N-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/85700/85654-06-4.png)
![Silanamine, 1,1,1-trimethyl-N-[(4-methylphenyl)methylene]-](http://img.cochemist.com/ccimg/85700/85654-06-4_b.png)