Co-reporter:Philipp Jöckle, Caroline Schweigert, Iris Lamparth, Norbert Moszner, Andreas-Neil Unterreiner, and Christopher Barner-Kowollik
Macromolecules November 28, 2017 Volume 50(Issue 22) pp:8894-8894
Publication Date(Web):November 9, 2017
DOI:10.1021/acs.macromol.7b01721
Five para-substituted monoacyltrimethylgermane derivatives, i.e., p-fluorobenzoyltrimethylgermane (pFBG, λmax = 405 nm), p-methoxybenzoyltrimethylgermane (pMBG, λmax = 397 nm), benzoyltrimethylgermane (pHBG, λmax = 409 nm), p-cyanobenzoyltrimethylgermane (pCBG, λmax = 425 nm), and p-nitrobenzoyltrimethylgermane (pNBG, λmax = 429 nm) are investigated via a combination of pulsed laser polymerization with subsequent electrospray ionization and mass spectrometry (PLP-ESI-MS) as well as femtosecond transient absorption spectroscopy. The relative initiation efficiencies of the initiating benzoyl radical fragments of pFBG, pMBG, and pHBG are determined using PLP-ESI-MS. The para-substituted derivatives with the electron-donating groups, pFBG and pMBG, display a factor 1.5 and 1.3, respectively, superior overall initiation efficiency compared to the unsubstituted pHBG. In contrast, the derivatives pCBG and pNBG carrying electron-withdrawing groups display only weak initiation behavior at a factor 4 higher total energy of ∼112 J (∼28 J for typical PLP experiments with pMBG, pFBG, and pHBG at ∼320 J and 90 000 pulses). The differences in the initiation efficiencies are representative for two classes of monoacyltrimethylgermane initiators, i.e., efficient initiators and weak initiators, each distinct in their specific radical cleavage mechanism. The efficient initiators pMBG, pFBG, and pHBG show an ultrafast intersystem crossing within 2–4 ps after pulse irradiation and subsequent formation of benzoyl and trimethylgermyl radical fragments. In contrast, the weak initiators pCBG and pNBG relax to the ground state after photoexcitation via a dominating ultrafast internal conversion (IC) within 13 and 2 ps, respectively, disallowing effective initiation under typical PLP conditions (∼320 J/pulse with 90 000 pulses resulting in ∼28 J total energy per sample). pCBG features weak initiation behavior additionally forming methyl and p-cyanobenzoyldimethylgermyl radicals at a factor 4 higher total energy of ∼112 J. Consistent with a considerably faster IC relaxation, pNBG features a factor 10 weaker monomer conversion than pCBG.
Co-reporter:Elena Frick, Caroline Schweigert, Benjamin B. Noble, Hanna A. Ernst, Andrea Lauer, Yu Liang, Dominik Voll, Michelle L. Coote, Andreas-Neil Unterreiner, and Christopher Barner-Kowollik
Macromolecules 2016 Volume 49(Issue 1) pp:80-89
Publication Date(Web):November 13, 2015
DOI:10.1021/acs.macromol.5b02336
The fundamental influence of the structure and substitution of radical photoinitiators was investigated via a trifold combination of pulsed-laser polymerization with subsequent electrospray-ionization mass spectrometry (PLP-ESI-MS), femtosecond transient absorption (fs-TA) spectroscopy, and quantum chemistry. For the first time, a library of benzoin-derived photoinitiators with varied substitution patterns was synthesized. In the PLP-ESI-MS study, different photoinitiators were compared pairwise in so-called cocktail experiments, enabling the direct comparison of their initiation efficiency. In the fs-TA experiments, the transient response was observed after UV excitation in the visible spectral region, allowing for a description of excited state dynamics, which was analyzed with the aid of TD-DFT calculations. Ab initio calculations were undertaken to determine the reactivity of the radical fragments generated from these photoinitiators and to quantify the influence of various substituents on the rate of addition to monomer. In summary, the influence of the substituent on the initiation efficiency, intersystem crossing (ISC) behavior, excited state dynamics, and the extinction coefficients were analyzed. Hence, relaxation pathways and reaction mechanisms were optimized to explain disparate initiation efficiencies of a wide range of newly designed photoinitiators with varying substitution patterns. The strongly divergent absorptivities of the different photoinitiators and their corresponding initiation efficiencies underline that the absorptivity of a molecule is by no means an unequivocal measure for its reactivity when excited at a specific wavelength. In fact, the most efficient initiators are governed by one nπ* singlet state with a very low extinction coefficient at the excitation wavelength and one or two triplet states with nπ* character.
Co-reporter:M. Klinger, C. Schenk, F. Henke, A. Clayborne, A. Schnepf and A.-N. Unterreiner
Chemical Communications 2015 vol. 51(Issue 61) pp:12278-12281
Publication Date(Web):03 Jul 2015
DOI:10.1039/C5CC04513D
Femtosecond pump–probe absorption spectroscopy in tetrahydrofuran solution has been used to investigate the dynamics of a metalloid cluster compound {Ge9[Si(SiMe3)3]3}−1. Upon UV photoexcitation, the transients in the near-infrared spectral region showed signatures reminiscent of excess electrons in THF (bound or quasi-free) whereas in the visible part excited state dynamics of the cluster complex dominates.
Co-reporter:Hanna A. Ernst, Thomas J. A. Wolf, Oliver Schalk, Núria González-García, Andrey E. Boguslavskiy, Albert Stolow, Matthias Olzmann, and Andreas-Neil Unterreiner
The Journal of Physical Chemistry A 2015 Volume 119(Issue 35) pp:9225-9235
Publication Date(Web):August 12, 2015
DOI:10.1021/acs.jpca.5b04900
The photolysis of o-nitrophenol (o-NP), a typical push–pull molecule, is of current interest in atmospheric chemistry as a possible source of nitrous acid (HONO). To characterize the largely unknown photolysis mechanism, the dynamics of the lowest lying excited singlet state (S1) of o-NP was investigated by means of femtosecond transient absorption spectroscopy in solution, time-resolved photoelectron spectroscopy (TRPES) in the gas phase and quantum chemical calculations. Evidence of the unstable aci-nitro isomer is provided both in the liquid and in the gas phase. Our results indicate that the S1 state displays strong charge transfer character, which triggers excited state proton transfer from the OH to the NO2 group as evidenced by a temporal shift of 20 fs of the onset of the photoelectron spectrum. The proton transfer itself is found to be coupled to an out-of-plane rotation of the newly formed HONO group, finally leading to a conical intersection between S1 and the ground state S0. In solution, return to S0 within 0.2–0.3 ps was monitored by stimulated emission. As a competitive relaxation channel, ultrafast intersystem crossing to the upper triplet manifold on a subpicosecond time scale occurs both in solution and in the gas phase. Due to the ultrafast singlet dynamics, we conclude that the much discussed HONO split-off is likely to take place in the triplet manifold.
Co-reporter:Dr. Effi Bätzner;Dr. Yu Liang;Caroline Schweigert;Dr. Andreas-Neil Unterreiner; Dr. Hans-Achim Wagenknecht
ChemPhysChem 2015 Volume 16( Issue 8) pp:1607-1612
Publication Date(Web):
DOI:10.1002/cphc.201500062
Abstract
The C-nucleoside based on the hydroxyquinoline ligand (Hq) is complementary to itself and forms stable Hq–Hq pairs in double-stranded DNA. These artificial Hq–Hq pairs may serve as artificial electron carriers for long-range photoinduced electron transfer in DNA, as elucidated by a combination of gel electrophoretic analysis of irradiated samples and time-resolved transient absorption spectroscopy. For this study, the Hq–Hq pair was combined with a DNA-based donor–acceptor system consisting of 6-N,N-dimethylaminopyrene conjugated to 2′-deoxyuridine as photoinducible electron donor, and methyl viologen attached to the 2′-position of uridine as electron acceptor. The Hq radical anion was identified in the time-resolved measurements and strand cleavage products support its role as an intermediate charge carrier. Hence, the Hq–Hq pair significantly enhances the electron hopping capability of DNA compared to natural DNA bases over long distances while keeping the self-assembly properties as the most attractive feature of DNA as a supramolecular architecture.
Co-reporter:Amer Baniodeh;Yu Liang;Christopher E. Anson;Nicola Magnani;Annie K. Powell;Simon Seyfferle;Michael Slota;Martin Dressel;Lapo Bogani;Karin Goß
Advanced Functional Materials 2014 Volume 24( Issue 40) pp:6280-6290
Publication Date(Web):
DOI:10.1002/adfm.201400336
We investigated the electronic properties of the molecular magnetic nanotoruses [FeIII 10LnIII 10(Me-tea)10(Me-teaH)10(NO3)10], examining the dependence on the lanthanide (Ln) of both the intra and intermolecular electronic channels. Using femtosecond absorption spectroscopy we show that the intramolecular electronic channels follow a three-step process, which involves vibrational cooling and crossing to shallow states, followed by recombination. A comparison with the energy gaps showed a relationship between trap efficiency and gaps, indicating that lanthanide ions create trap states to form excitons after photo-excitation. Using high-resistance transport measurements and scaling techniques, we investigated the intermolecular transport, demonstrating the dominant role of surface-limited transport channels and the presence of different types of charge traps. The intermolecular transport properties can be rationalized in terms of a hopping model, and a connection is provided to the far-IR spectroscopic properties. Comparison between intra and intermolecular processes highlights the role of the excited electronic states and the recombination processes, showing the influence of Kramers parity on the overall mobility.
Co-reporter:T. J. A. Wolf, T. S. Kuhlman, O. Schalk, T. J. Martínez, K. B. Møller, A. Stolow and A.-N. Unterreiner
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 23) pp:11770-11779
Publication Date(Web):30 Apr 2014
DOI:10.1039/C4CP00977K
Progress in our understanding of ultrafast light-induced processes in molecules is best achieved through a close combination of experimental and theoretical approaches. Direct comparison is obtained if theory is able to directly reproduce experimental observables. Here, we present a joint approach comparing time-resolved photoelectron spectroscopy (TRPES) with ab initio multiple spawning (AIMS) simulations on the MS-MR-CASPT2 level of theory. We disentangle the relationship between two phenomena that dominate the immediate molecular response upon light absorption: a spectrally dependent delay of the photoelectron signal and an induction time prior to excited state depopulation in dynamics simulations. As a benchmark molecule, we have chosen hexamethylcyclopentadiene, which shows an unprecedentedly large spectral delay of (310 ± 20) fs in TRPES experiments. For the dynamics simulations, methyl groups were replaced by “hydrogen atoms” having mass 15 and TRPES spectra were calculated. These showed an induction time of (108 ± 10) fs which could directly be assigned to progress along a torsional mode leading to the intersection seam with the molecular ground state. In a stepladder-type approach, the close connection between the two phenomena could be elucidated, allowing for a comparison with other polyenes and supporting the general validity of this finding for their excited state dynamics. Thus, the combination of TRPES and AIMS proves to be a powerful tool for a thorough understanding of ultrafast excited state dynamics in polyenes.
Co-reporter:T. J. A. Wolf, O. Schalk, R. Radloff, G. Wu, P. Lang, A. Stolow and A.-N. Unterreiner
Physical Chemistry Chemical Physics 2013 vol. 15(Issue 18) pp:6673-6683
Publication Date(Web):11 Feb 2013
DOI:10.1039/C3CP44295K
The photoinduced dynamics of the fully halogenated cyclopentadienes C5Cl6 and C5Br6 have been investigated in solution and gas phase by femtosecond time-resolved spectroscopy. Both in solution and in gas phase, homolytic dissociation into a halogen radical and a C5X5 (X = Cl, Br) radical was observed. In liquid phase, solvent-dependent formation of charge transfer complexes between geminate radicals was observed for the first time. These complexes were found to be surprisingly stable and offered the opportunity to follow the dynamics of specific radical pairs. In the case of C5Cl6 in trichloroethanol, a reaction of the chlorine radical with molecules from the solvent cage was observed.
Co-reporter:Dr. Yu Liang;Maximilian Bradler;Dr. Melanie Klinger;Dr. Oliver Schalk;Mihaela Carmen Balaban;Dr. Teodor Silviu Balaban;Dr. Eberhard Riedle;Dr. Andreas-Neil Unterreiner
ChemPlusChem 2013 Volume 78( Issue 10) pp:1244-1251
Publication Date(Web):
DOI:10.1002/cplu.201300143
Abstract
Studying the relaxation pathways of porphyrins and related structures upon light absorption is crucial to understand the fundamental processes of light harvesting in biosystems and many applications. Herein, we show by means of transient absorption studies, following Q- and Soret-band excitation, and ab initio calculations on meso-tetraphenylporphyrinato magnesium(II) (MgTPP) and meso-tetraphenylporphyrinato cadmium(II) (CdTPP) that electronic relaxation following Soret-band excitation of porphyrins with a heavy central atom is mediated by a hitherto disregarded dark state. This accounts for an increased rate of internal conversion. The dark state originates from an orbital localized at the central nitrogen atoms and its energy continuously decreases along the series from magnesium to zinc to cadmium to below 2.75 eV for CdTPP dissolved in tetrahydrofuran. Furthermore, we are able to directly trace fast intersystem crossing in the cadmium derivative, which takes place within (110±20) ps.
Co-reporter:Dr. Thomas Ehrenschwender;Dr. Yu Liang;Dr. Andreas-Neil Unterreiner; Dr. Hans-Achim Wagenknecht;Dr. Thomas J. A. Wolf
ChemPhysChem 2013 Volume 14( Issue 6) pp:1197-1204
Publication Date(Web):
DOI:10.1002/cphc.201200924
Abstract
A new donor-DNA-acceptor system has been synthesized containing Nile red-modified 2′-deoxyuridine as charge donor and 6-N,N-dimethylaminopyrene-modified 2′-deoxyuridine as acceptor to investigate the charge transfer in DNA duplexes using fluorescence spectroscopy and time-resolved femtosecond pump-probe techniques. Fluorescence quenching experiments revealed that the quenching efficiency of Nile red depends on two components: 1) the presence of a charge acceptor and 2) the number of intervening CG and AT base pairs between donor and acceptor. Surprisingly, the quenching efficiency of two base pairs (73 % for CG and the same for AT) is higher than that for one base pair (68 % for CG and 37 % for AT), while at a separation of three base pairs less than 10 % quenching is observed. A comparison with the results of time-resolved measurements revealed a correlation between quenching efficiency and the first ultrafast time constant suggesting that quenching proceeds via a charge transfer from the donor to the acceptor. All transients are satisfactorily described with two decays: a rapid charge transfer with 600 fs (∼1012 s−1) that depends strongly and in a non-linear fashion on the distance between donor and acceptor, and a slower time constant of a few picoseconds (∼1011 s−1) with weak distance dependence. A third time constant on a nanosecond time scale represents the fluorescence lifetime of the donor molecule. According to these results and time-dependent density functional theory (TDDFT) calculations a combination of single-step superexchange and multistep hopping mechanisms can be proposed for this short-range charge transfer. Furthermore, significantly less quenching efficiency and slower charge transfer rates at very short distances indicate that the direct interaction between donor and acceptor leads to a local structural distortion of DNA duplexes which may provide some uncertainty in identifying the charge transfer rates in short-range systems.
Co-reporter:Thomas J. A. Wolf, Dominik Voll, Christopher Barner-Kowollik, and Andreas-Neil Unterreiner
Macromolecules 2012 Volume 45(Issue 5) pp:2257-2266
Publication Date(Web):February 28, 2012
DOI:10.1021/ma202673q
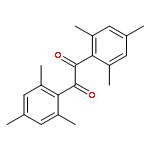
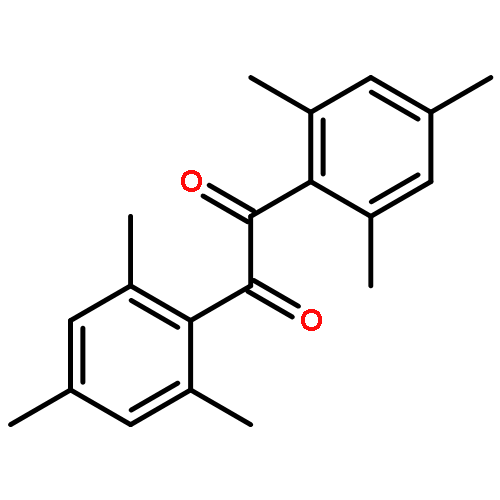
The excited states and dynamics of the three triplet radical photoinitiators benzoin (2-hydroxy-1,2-diphenylethanone, Bz), 2,4,6-trimethylbenzoin (2-hydroxy-1-mesityl-2-phenylethanone, TMB), and mesitil (1,2-bis(2,4,6-trimethylphenyl)-1,2-ethanedione, Me)—employed in our previous studies for quantifying net initiation efficiencies in pulsed laser polymerization with methacrylate monomers [Voll, D.; Junkers, T.; Barner-Kowollik, C. Macromolecules2011, 44, 2542–2551]—are investigated via both femtosecond transient absorption (TA) spectroscopy and density functional theory (DFT) methods to elucidate the underlying mechanisms causing different initiation efficiencies when excited at 351 nm. Bz and TMB are found to have very similar properties in the calculated excited states as well as in the experimentally observed dynamics. After excitation into the first excited singlet state (S1) Bz and TMB undergo rapid intersystem crossing (ISC). The ISC can compete with ultrafast internal conversion (IC) processes because an excited triplet state (Tn) of nearly the same energy is present in both cases. ISC is therefore the dominating depopulation channel of S1, and subsequent α-cleavage to produce radicals takes place on the picosecond time scale. In contrast, Me is excited into the second excited singlet state (S2). In this case no isoenergetic triplet state is available, which inhibits ISC from competing with ultrafast deactivation processes. ISC is therefore assigned to be a minor deactivation channel in Me. Employing these findings, quantitative photoinitiation efficiency relations of Bz, TMB, and Me obtained by pulsed laser polymerization can be directly correlated with the relative TA intensities found in the femtosecond experiments. The ISC efficiency is thus a critical parameter for evaluating the overall photoinitiation efficiency and demonstrates that the employment of the herein presented method represents a powerful tool for attaining a quantitative picture on the suitability of a photoinitiator.
Co-reporter:Frank Hennrich ; Manfred M. Kappes ; Melanie Klinger
The Journal of Physical Chemistry C 2011 Volume 115(Issue 48) pp:23711-23717
Publication Date(Web):October 26, 2011
DOI:10.1021/jp2075176
Time-resolved two color pump/probe spectroscopy was used to unravel the dynamics of ultrafast decay occurring upon population of the first optical bright excitonic level (E11) in quasi-monodispersed, polymer-wrapped, single-walled (9,7)-carbon nanotubes (SWNTs) in toluene at room temperature. After resonant E11 excitation, transfer of population to at least one optically dark level near E11 was observed to take place within the first picosecond. In addition, phonon-assisted E11-excitation led to transients similar to those observed upon resonant E11-excitation indicating ultrafast vibrational relaxation convoluted with the temporal resolution of 60 fs.
Co-reporter:Chandrasekhar Nese and Andreas-Neil Unterreiner
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 8) pp:1698-1708
Publication Date(Web):18 Jan 2010
DOI:10.1039/B916799B
The number of investigations of room temperature ionic liquids (RTILs) has been developing rapidly in recent years, e.g., due to their potential use as “green solvents” in many industrial applications. A large body of data has been accumulated on physico-chemical properties including ultrafast dynamics of these liquids. This review deals both with the generation and the subsequent relaxation dynamics of selected transient species following photolysis or pulse radiolysis of RTILs with special emphasis on the exceptional character of imidazolium based systems. Although considerable progress has been made, the understanding of the photochemistry of ionic liquids is just at its embryonic stage. Nevertheless, a brief comparison with high temperature ionic liquids (HTILs) such as alkali metal doped alkali halide mixtures already reveals important differences.
Co-reporter:Oliver Schalk and Andreas N. Unterreiner
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 3) pp:655-666
Publication Date(Web):18 Nov 2009
DOI:10.1039/B913360G
An extensive analysis of transient anisotropy is presented including an ansatz to describe the temporal evolution of anisotropy in multiphoton experiments in the limit of Brownian motion. For the general case, this evolution is described by means of a step model interpolating between collision-free reorientation and Brownian diffusion for different geometries. The presented ansatz is able to calculate the time dependence of the anisotropy for symmetric top molecules. This dependence is shown to be in third order with respect to the solvent–solute interaction irrespective of the molecular geometry. Differences to former models are worked out and an extension to rotational coherence effects is given. Finally, the influence of collisions on the anisotropy decay is modeled by Monte-Carlo simulations allowing for a variation of angular correlation and energy transfer.
Co-reporter:N. Chandrasekhar, O. Schalk and A.-N. Unterreiner
The Journal of Physical Chemistry B 2008 Volume 112(Issue 49) pp:15718-15724
Publication Date(Web):November 12, 2008
DOI:10.1021/jp804861z
Femtosecond pump−probe absorption spectroscopy was employed to investigate ultrafast dynamics in various room temperature ionic liquids (RTILs) based on imidazolium cations, i.e., 1,3-dimethylimidazolium iodide ([DMIM]I), 1-butyl-3-methylimidazolium iodide ([BMIM]I), 1-hexyl-3-methylimidazolium iodide ([HMIM]I), 1-hexyl-3-methylimidazolium chloride ([HMIM]Cl), and 1-methyl-3-octylimidazolium chloride ([MOIM]Cl). Immediately after photoexcitation, an induced absorption was observed at various probe wavelengths (555−1556 nm). Afterward, the decay of the induced absorption was found to be independent of the alkyl chain length and viscosity of the ionic liquids. Two alternative mechanisms were proposed to explain the dynamics. In a first scenario excess electrons are generated through one-photon photodetachment of halides analogous to aqueous halide photodetachment. The dynamics in this case were analyzed with the help of a competing kinetic model proposed for geminate recombination in aqueous chloride photodetachment. Alternatively, imidazolium cations may be subject to photoionization. The transient NIR absorption can then be assigned to imidazolium dimer radical cations and/or excess electrons which may be formed upon association of imidazolium radicals with their parent cations. Both scenarios suggest that a thorough explanation of the ultrafast dynamics probably requires the implication of cooperative effects in the ionic liquids upon photoexcitation.
Co-reporter:M. Klinger, C. Schenk, F. Henke, A. Clayborne, A. Schnepf and A.-N. Unterreiner
Chemical Communications 2015 - vol. 51(Issue 61) pp:NaN12281-12281
Publication Date(Web):2015/07/03
DOI:10.1039/C5CC04513D
Femtosecond pump–probe absorption spectroscopy in tetrahydrofuran solution has been used to investigate the dynamics of a metalloid cluster compound {Ge9[Si(SiMe3)3]3}−1. Upon UV photoexcitation, the transients in the near-infrared spectral region showed signatures reminiscent of excess electrons in THF (bound or quasi-free) whereas in the visible part excited state dynamics of the cluster complex dominates.
Co-reporter:T. J. A. Wolf, T. S. Kuhlman, O. Schalk, T. J. Martínez, K. B. Møller, A. Stolow and A.-N. Unterreiner
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 23) pp:NaN11779-11779
Publication Date(Web):2014/04/30
DOI:10.1039/C4CP00977K
Progress in our understanding of ultrafast light-induced processes in molecules is best achieved through a close combination of experimental and theoretical approaches. Direct comparison is obtained if theory is able to directly reproduce experimental observables. Here, we present a joint approach comparing time-resolved photoelectron spectroscopy (TRPES) with ab initio multiple spawning (AIMS) simulations on the MS-MR-CASPT2 level of theory. We disentangle the relationship between two phenomena that dominate the immediate molecular response upon light absorption: a spectrally dependent delay of the photoelectron signal and an induction time prior to excited state depopulation in dynamics simulations. As a benchmark molecule, we have chosen hexamethylcyclopentadiene, which shows an unprecedentedly large spectral delay of (310 ± 20) fs in TRPES experiments. For the dynamics simulations, methyl groups were replaced by “hydrogen atoms” having mass 15 and TRPES spectra were calculated. These showed an induction time of (108 ± 10) fs which could directly be assigned to progress along a torsional mode leading to the intersection seam with the molecular ground state. In a stepladder-type approach, the close connection between the two phenomena could be elucidated, allowing for a comparison with other polyenes and supporting the general validity of this finding for their excited state dynamics. Thus, the combination of TRPES and AIMS proves to be a powerful tool for a thorough understanding of ultrafast excited state dynamics in polyenes.
Co-reporter:Oliver Schalk and Andreas N. Unterreiner
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 3) pp:NaN666-666
Publication Date(Web):2009/11/18
DOI:10.1039/B913360G
An extensive analysis of transient anisotropy is presented including an ansatz to describe the temporal evolution of anisotropy in multiphoton experiments in the limit of Brownian motion. For the general case, this evolution is described by means of a step model interpolating between collision-free reorientation and Brownian diffusion for different geometries. The presented ansatz is able to calculate the time dependence of the anisotropy for symmetric top molecules. This dependence is shown to be in third order with respect to the solvent–solute interaction irrespective of the molecular geometry. Differences to former models are worked out and an extension to rotational coherence effects is given. Finally, the influence of collisions on the anisotropy decay is modeled by Monte-Carlo simulations allowing for a variation of angular correlation and energy transfer.
Co-reporter:Chandrasekhar Nese and Andreas-Neil Unterreiner
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 8) pp:NaN1708-1708
Publication Date(Web):2010/01/18
DOI:10.1039/B916799B
The number of investigations of room temperature ionic liquids (RTILs) has been developing rapidly in recent years, e.g., due to their potential use as “green solvents” in many industrial applications. A large body of data has been accumulated on physico-chemical properties including ultrafast dynamics of these liquids. This review deals both with the generation and the subsequent relaxation dynamics of selected transient species following photolysis or pulse radiolysis of RTILs with special emphasis on the exceptional character of imidazolium based systems. Although considerable progress has been made, the understanding of the photochemistry of ionic liquids is just at its embryonic stage. Nevertheless, a brief comparison with high temperature ionic liquids (HTILs) such as alkali metal doped alkali halide mixtures already reveals important differences.
Co-reporter:T. J. A. Wolf, O. Schalk, R. Radloff, G. Wu, P. Lang, A. Stolow and A.-N. Unterreiner
Physical Chemistry Chemical Physics 2013 - vol. 15(Issue 18) pp:NaN6683-6683
Publication Date(Web):2013/02/11
DOI:10.1039/C3CP44295K
The photoinduced dynamics of the fully halogenated cyclopentadienes C5Cl6 and C5Br6 have been investigated in solution and gas phase by femtosecond time-resolved spectroscopy. Both in solution and in gas phase, homolytic dissociation into a halogen radical and a C5X5 (X = Cl, Br) radical was observed. In liquid phase, solvent-dependent formation of charge transfer complexes between geminate radicals was observed for the first time. These complexes were found to be surprisingly stable and offered the opportunity to follow the dynamics of specific radical pairs. In the case of C5Cl6 in trichloroethanol, a reaction of the chlorine radical with molecules from the solvent cage was observed.







![2-[4-(DIMETHYLAMINO)PHENYL]-2-HYDROXY-1-PHENYLETHANONE](http://img.cochemist.com/ccimg/33500/33458-29-6.png)
![2-[4-(DIMETHYLAMINO)PHENYL]-2-HYDROXY-1-PHENYLETHANONE](http://img.cochemist.com/ccimg/33500/33458-29-6_b.png)

![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3.png)
![5H-Benzo[a]phenoxazin-5-one,9-(diethylamino)-](http://img.cochemist.com/ccimg/7400/7385-67-3_b.png)
![Ethanone,1-[4-(dimethylamino)phenyl]-2-hydroxy-2-phenyl-](http://img.cochemist.com/ccimg/6400/6317-85-7.png)
![Ethanone,1-[4-(dimethylamino)phenyl]-2-hydroxy-2-phenyl-](http://img.cochemist.com/ccimg/6400/6317-85-7_b.png)