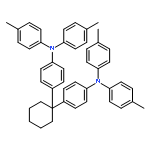
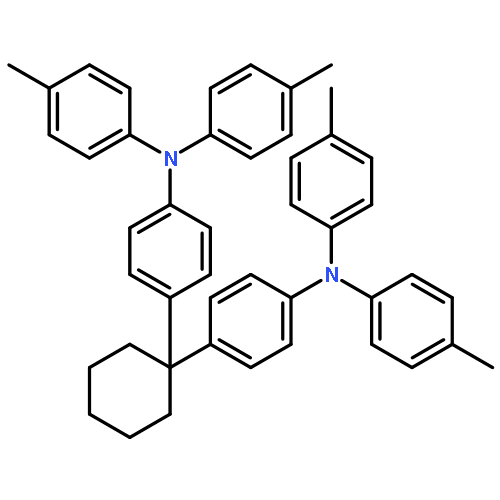
Co-reporter:C. Adrian Figg;Ashton N. Bartley;Tomohiro Kubo;Bryan S. Tucker;Brent S. Sumerlin
Polymer Chemistry (2010-Present) 2017 vol. 8(Issue 16) pp:2457-2461
Publication Date(Web):2017/04/18
DOI:10.1039/C7PY00225D
This report describes the efficient preparation of ω,ω-heterodifunctionalized polymers and polymer bioconjugates under mild conditions using a recently introduced reagent, benzotrifuranone (BTF). Demonstrated is how BTF enables introduction of differentially “clickable” functional groups (e.g., alkenes and alkynes) to monomethyl ether poly(ethylene glycol) amine at ambient temperature using near-stoichiometric amounts of reagents, in contrast to conventional polymer heterofunctionalization approaches that may require high temperatures, significant excesses of reagents, and/or numerous synthetic steps. Showcasing the methodology is facile access to an ω,ω-heterodifunctional polymer bearing a fluorescent (coumarin) dye and biotin. Found, in addition to avidin binding by the polymer bioconjugate, is the unexpected disruption of avidin tetramer formation.
Co-reporter:Renan B. Ferreira;Jose M. Figueroa;Danielle E. Fagnani;Khalil A. Abboud
Chemical Communications 2017 vol. 53(Issue 69) pp:9590-9593
Publication Date(Web):2017/08/24
DOI:10.1039/C7CC04505K
Reported here is the first synthesis, X-ray crystal structure, and derivatization of benzotrifuran (BTFuran). Single crystal X-ray analysis of BTFuran shows a tight hexagonal packing stabilized by π-stacking interactions and C–H···O contacts. α-Lithiation of BTFuran enables the preparation of reactive intermediates suitable for cross-coupling reactions, allowing access to representative BTFuran-containing π-conjugated systems.
Co-reporter:Matthew B. Baker, Renan B. Ferreira, Jonathan Tasseroul, Andrew J. Lampkins, Alexandre Al Abbas, Khalil A. Abboud, and Ronald K. Castellano
The Journal of Organic Chemistry 2016 Volume 81(Issue 19) pp:9279-9288
Publication Date(Web):August 31, 2016
DOI:10.1021/acs.joc.6b01867
Benzotrifuranone (BTF), bearing three symmetry-equivalent lactone rings, is unique in its ability to undergo highly selective and sequential aminolysis reactions in one-pot to afford multifunctionalized molecules (>80% overall yield). New insight into this behavior is presented through kinetics measurements (by stopped-flow IR spectroscopy), X-ray crystal structure analysis, quantum chemical calculations, and comparison of BTF to other benzoate esters, including its ring expanded congener benzotripyranone (BTP). While the structure–property investigation confirms stepwise electronic/inductive lactone deactivation for both BTF and BTP, the unusually fast and selective aminolysis of BTF is only fully explained through synergistic ring strain effects. Experimental signatures of the significant ring strain of BTF (∼28 kcal mol–1 based on DFT calculations vs 17 kcal mol–1 for BTP) include its high lactone carbonyl stretching energy (1821 cm–1 in acetonitrile vs 1777 cm–1 for BTP) and bond length alternation within its benzenoid ring. While ring strain is relieved upon the sequential aminolysis of both BTF and BTP, it is only for the former that a ring strain gradient is established that contributes to the stepwise aminolysis rate differences and enhanced selectivity. The work shows how a combination of electronic effects and ring strain can underpin the design of small molecules capable of stepwise functionalization, of which there are notably few examples.
Co-reporter:Danielle E. Fagnani;Michael J. Meese Jr.;Dr. Khalil A. Abboud ;Dr. Ronald K. Castellano
Angewandte Chemie International Edition 2016 Volume 55( Issue 36) pp:10726-10731
Publication Date(Web):
DOI:10.1002/anie.201605286
Abstract
[2.2]paracyclophane (pCp), unlike many π-building blocks, has been virtually unexplored in supramolecular constructs. Reported here is the synthesis and characterization of the first pCp derivatives capable of programmed self-assembly into extended cofacial π-stacks in solution and the solid state. The design employs transannular (intramolecular) hydrogen bonds (H-bonds), hitherto unstudied in pCps, between pseudo-ortho-positioned amides of a pCp-4,7,12,15-tetracarboxamide (pCpTA) to preorganize the molecules for intermolecular H-bonding with π-stacked neighbors. X-ray crystallography confirms the formation of homochiral, one-dimensional pCpTA stacks helically laced with two H-bond strands. The chiral sense is dictated by the planar chirality (Rp or Sp) of the pCpTA monomers. A combination of NMR, IR, and UV/Vis studies confirms the formation of the first supramolecular pCp polymers in solution.
Co-reporter:Danielle E. Fagnani;Michael J. Meese Jr.;Dr. Khalil A. Abboud ;Dr. Ronald K. Castellano
Angewandte Chemie 2016 Volume 128( Issue 36) pp:10884-10889
Publication Date(Web):
DOI:10.1002/ange.201605286
Abstract
[2.2]paracyclophane (pCp), unlike many π-building blocks, has been virtually unexplored in supramolecular constructs. Reported here is the synthesis and characterization of the first pCp derivatives capable of programmed self-assembly into extended cofacial π-stacks in solution and the solid state. The design employs transannular (intramolecular) hydrogen bonds (H-bonds), hitherto unstudied in pCps, between pseudo-ortho-positioned amides of a pCp-4,7,12,15-tetracarboxamide (pCpTA) to preorganize the molecules for intermolecular H-bonding with π-stacked neighbors. X-ray crystallography confirms the formation of homochiral, one-dimensional pCpTA stacks helically laced with two H-bond strands. The chiral sense is dictated by the planar chirality (Rp or Sp) of the pCpTA monomers. A combination of NMR, IR, and UV/Vis studies confirms the formation of the first supramolecular pCp polymers in solution.
Co-reporter:Nathan T. Shewmon;Davita L. Watkins;Johan F. Galindo;Raghida Bou Zerdan;Jihua Chen;Jong Keum;Adrian E. Roitberg;Jiangeng Xue
Advanced Functional Materials 2015 Volume 25( Issue 32) pp:5166-5177
Publication Date(Web):
DOI:10.1002/adfm.201501815
For organic photovoltaic (OPV) cells based on the bulk heterojunction (BHJ) structure, it remains challenging to rationally control the degree of phase separation and percolation within blends of donors and acceptors to secure optimal charge separation and transport. Reported is a bottom-up, supramolecular approach to BHJ OPVs wherein tailored hydrogen bonding (H-bonding) interactions between π-conjugated electron donor molecules encourage formation of vertically aligned donor π-stacks while simultaneously suppressing lateral aggregation; the programmed arrangement facilitates fine mixing with fullerene acceptors and efficient charge transport. The approach is illustrated using conventional linear or branched quaterthiophene donor chromophores outfitted with terminal functional groups that are either capable or incapable of self-complementary H-bonding. When applied to OPVs, the H-bond capable donors yield a twofold enhancement in power conversion efficiency relative to the comparator systems, with a maximum external quantum efficiency of 64%. H-bond promoted assembly results in redshifted absorption (in neat films and donor:C60 blends) and enhanced charge collection efficiency despite disparate donor chromophore structure. Both features positively impact photocurrent and fill factor in OPV devices. Film structural characterization by atomic force microscopy, transmission electron microscopy, and grazing incidence wide angle X-ray scattering reveals a synergistic interplay of lateral H-bonding interactions and vertical π-stacking for directing the favorable morphology of the BHJ.
Co-reporter:Xueying Zhao, Davita L. Watkins, Johan F. Galindo, Nathan T. Shewmon, Adrian E. Roitberg, Jiangeng Xue, Ronald K. Castellano, Scott S. Perry
Organic Electronics 2015 Volume 19() pp:61-69
Publication Date(Web):April 2015
DOI:10.1016/j.orgel.2015.01.022
•Tailored H-bonds were used to control supramolecular assembly of donor molecules.•STM studies confirmed the formation of ordered H-bonded trimer rosettes on Au(1 1 1).•DFT calculations facilitated the structural understanding of H-bonded assembly.•Observed templated growth of C60 on ordered monolayer of H-bonded donor molecules.We previously reported that a branched quaterthiophene donor chromophore functionalized with a phthalhydrazide hydrogen bonding (H-bonding) unit (MeBQPH) gives twofold more efficient bulk heterojunction organic solar cells (with C60 acceptors) compared to a nearly identical donor incapable of H-bonding (MeBQPME). Here, scanning tunneling microscopy (STM) studies confirm the formation of MeBQPH trimer rosettes on Au(1 1 1) through phthalhydrazide H-bonding interactions that are sufficiently strong to compete with adsorbate/substrate interactions. The MeBQPME comparator molecule void of hydrogen bonding functionality does not similarly assemble on the metal surface. Complementary density functional theory (DFT) calculations facilitate a structural understanding of the MeBQPH donor assemblies and their strong stabilization through formation of six hydrogen bonds. STM studies also reveal the templated growth of C60 on ordered MeBQPH monolayers with C60 molecules preferentially occupying the threefold interstitial sites of the MeBQPH monolayer. This work supports the idea that H-bonding interactions can be used to control the morphology of organic donor–acceptor blends to potentially create efficient and stable bulk heterojunction photovoltaic devices.
Co-reporter:Raghida Bou Zerdan, Pamela Cohn, Egle Puodziukynaite, Matthew B. Baker, Maud Voisin, Céline Sarun, and Ronald K. Castellano
The Journal of Organic Chemistry 2015 Volume 80(Issue 3) pp:1828-1840
Publication Date(Web):January 12, 2015
DOI:10.1021/jo502773g
The molecular recognition properties of the nucleobases instruct the formation of complex three-dimensional architectures in natural and synthetic systems; relatively unexplored is their use as building blocks for π-conjugated materials where they might mutually tune electronic and supramolecular structures. Toward this goal, an introductory set (1a–d and 2a–d) of six purine-terminated and two pyrimidine-terminated π-conjugated oligomers has been synthesized and used to develop experimental electronic and photophysical structure–property trends. Unlike 2,2′:5′,2″-terthiophene (TTT) derivatives 2a–d, intramolecular charge transfer dominates oligomers 1a–d bearing a 4,7-bisthienylbenzothiadiazole (TBT) spacer due to the strong electron-accepting ability of its benzothiadiazole (BTD) ring. The resulting donor–acceptor–donor systems feature lower HOMO–LUMO gaps than the terthiophene-linked nucleobases (ΔEg ∼ 1.8 eV vs 2.4 eV based on electrochemical measurements), and the lowest so far for π-conjugated molecules that include nucleobases within the π-framework. Experiments reveal a dependence of photophysical and electronic structure on the nature of the nucleobase and are in good agreement with theoretical calculations performed at the B3LYP/6-31+G** level. Overall, the results show how nucleobase heterocycles can be installed within π-systems to tune optical and electronic properties. Future work will evaluate the consequences of these information-rich components on supramolecular π-conjugated structure.
Co-reporter:Raghida Bou Zerdan;Nathan T. Shewmon;Yu Zhu;John P. Mudrick;Kyle J. Chesney;Jiangeng Xue
Advanced Functional Materials 2014 Volume 24( Issue 38) pp:5993-6004
Publication Date(Web):
DOI:10.1002/adfm.201401030
Three stereochemically pure isomers and two isomeric mixtures of a solution-processable diketopyrrolopyrrole-containing oligothiophene (SMDPPEH) have been used to study the effect of 2-ethylhexyl solubilizing group stereochemistry on the film morphology and bulk heterojunction (BHJ) solar cell characteristics of small molecule organic photovoltaics. The different SMDPPEH stereoisomer compositions exhibit nearly identical optoelectronic properties in the molecularly dissolved state, as well as in amorphous films blended with PCBM. However, for films in which SMDPPEH crystallization is induced by thermal annealing, significant differences in molecular packing between the different stereoisomer formulations are observed. These differences are borne out in photovoltaic device characteristics for which unannealed devices show very similar behavior, while after annealing the RR- and SS-SMDPPEH enantiomers show blue-shifted peak EQE relative to the SMDPPEH isomer mixtures. Unannealed devices made from the most crystalline stereoisomer, meso RS-SMDPPEH, are not completely amorphous, and show improved photocurrent generation as a result. Unlike the other compounds, after thermal annealing the RS-SMDPPEH devices show reduced device performance. The results reveal that the chirality of commonly used 2-ethylhexyl solubilizing chains can have a significant effect on the morphology, absorption, and optimum processing conditions of small molecule organic thin films used as photovoltaic device active layers.
Co-reporter:Benjamin M. Schulze, Nathan T. Shewmon, Jing Zhang, Davita L. Watkins, John P. Mudrick, Weiran Cao, Raghida Bou Zerdan, Anthony J. Quartararo, Ion Ghiviriga, Jiangeng Xue and Ronald K. Castellano
Journal of Materials Chemistry A 2014 vol. 2(Issue 5) pp:1541-1549
Publication Date(Web):04 Dec 2013
DOI:10.1039/C3TA13529B
Reported is a systematic molecular structure–property relationship study to evaluate the consequences of dedicated H-bonding interactions between molecular electron donors on molecular assembly, absorption, charge collection, and performance in small-molecule bulk heterojunction organic photovoltaic devices. Three families of branched quaterthiophene donor chromophores have been synthesized with members that share nearly identical electronic and optical properties in the molecularly dispersed state but are either capable or incapable of self-association by hydrogen bonding (H-bonding). Phthalhydrazide-functionalized quaterthiophenes are H-bond “active” and show signatures of H-bond promoted assembly in solution (by 1H NMR) and in both neat and blended (with C60) films (by IR). Compared to control compounds with H-bonding “turned off”, the H-bonded derivatives show red-shifted thin film absorption (neat and as blends with C60), different colors as bulk solids, and increased decomposition and melt temperatures. Photovoltaic devices made from blends of H-bonded donor molecules with C60 as the electron acceptor show improved charge collection length and external quantum efficiency resulting in a more than two-fold enhancement in power conversion efficiency relative to non-H-bonding controls, from 0.49% to 1.04%. We anticipate this approach could be generalized to include other donor chromophores with lower optical gap to harvest more longer-wavelength photons and achieve higher power conversion efficiencies.
Co-reporter:Benjamin M. Schulze, Davita L. Watkins, Jing Zhang, Ion Ghiviriga and Ronald K. Castellano
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 40) pp:7932-7936
Publication Date(Web):28 Aug 2014
DOI:10.1039/C4OB01373E
Reported is characterization of the self-assembly of π-conjugated oligomers, molecules studied recently in photovoltaic devices, using variable temperature diffusion ordered spectroscopy (VT-DOSY). Iterative fitting of diffusion coefficient versus temperature data to a modified Stokes–Einstein equation, molecular modelling, and comparison to non-assembling model compounds, has allowed estimation of assembly size, shape, and molecularity.
Co-reporter:Yixing Yang, Pamela Cohn, Sang-Hyun Eom, Khalil A. Abboud, Ronald K. Castellano and Jiangeng Xue
Journal of Materials Chemistry A 2013 vol. 1(Issue 16) pp:2867-2874
Publication Date(Web):18 Feb 2013
DOI:10.1039/C3TC00734K
We report efficient ultraviolet (UV)-violet organic light-emitting devices (OLEDs) based on highly fluorescent donor–acceptor purine molecules, which can generate tunable emission from 350 nm to 450 nm in solution by using different electron donor and acceptor arrangements on the heterocycles as reported previously. Here, external quantum efficiencies (EQEs) up to ηEQE = 1.6% are achieved for the multilayer OLEDs based on purine 2, with UV emission peaked at 393 nm, as compared to previously reported purine 1 based OLEDs with ηEQE = 3.1% and peak emission at 433 nm. The efficiencies of the OLEDs based on the two purine molecules are among the highest reported to date with emission peak wavelengths below 450 nm. By using a range of charge transport and host materials, we show that appropriate energy level alignment in multilayer OLED devices is imperative to achieve UV emission and prevent undesired emission from other layers or interfaces.
Co-reporter:Matthew B. Baker, Ion Ghiviriga and Ronald K. Castellano
Chemical Science 2012 vol. 3(Issue 4) pp:1095-1099
Publication Date(Web):20 Dec 2011
DOI:10.1039/C2SC00943A
Synthetic access to discrete multifunctionalized molecules remains a challenge but is critical for continued advances in materials science, catalysis, and chemical biology. Here, the aminolysis of electronically coupled lactones is introduced as an approach to achieve molecular multifunctionalization efficiently, under mild conditions, and without protecting groups or by-products. The one-pot sequential aminolysis of benzotrifuranone, a C3h-symmetric trilactone, to produce a trifunctionalized target in ∼85% yield over one day is highlighted.
Co-reporter:Ronald K. Castellano;Yan Li;Edwin A. Homan;Andrew J. Lampkins;Iris V. Marín ;Khalil A. Abboud
European Journal of Organic Chemistry 2012 Volume 2012( Issue 24) pp:4483-4492
Publication Date(Web):
DOI:10.1002/ejoc.201200438
Abstract
Seven-membered intramolecular hydrogen bonding (7MHB) arrangements involving phenolic hydroxyl group donors have been investigated through Cambridge Structural Database (CSD) and literature mining, and by the characterization of model compounds. The CSD reveals the numerous H-bond accepting functional groups that can participate in 7MHB when they are proximal to a phenolic OH, including alcohols, amides, amines, ethers, N-containing heterocycles, ketones, N-oxides, phosphates, and phosphane oxides. The HB contacts are defined by Ophenol···N and Ophenol···O distances of ca. 2.7 Å, and two dihedral angles that fall within the range of typical peptide γ-turns. Two of the identified 7MHB motifs have been readily mapped onto the phloroglucinol (1,3,5-trihydroxybenzene) scaffold to provide eight model compounds; intramolecular hydrogen bonding involving all three hydroxyl groups is shown to persist in solution and the solid state for the compounds. Intramolecular 7MHB enforces nonplanar conformations that should be useful when designing molecular hosts and catalysts.
Co-reporter:Yixing Yang, Pamela Cohn, Aubrey L. Dyer, Sang-Hyun Eom, John R. Reynolds, Ronald K. Castellano and Jiangeng Xue
Chemistry of Materials 2010 Volume 22(Issue 12) pp:3580
Publication Date(Web):May 25, 2010
DOI:10.1021/cm100407n
Co-reporter:Yan Li, Andrew J. Lampkins, Matthew B. Baker, Bobby G. Sumpter, Jingsong Huang, Khalil A. Abboud and Ronald K. Castellano
Organic Letters 2009 Volume 11(Issue 19) pp:4314-4317
Publication Date(Web):August 28, 2009
DOI:10.1021/ol901631n
Functionalized benzotrifurans can be accessed in one efficient acylation step from previously unreported benzotrifuranone. DFT calculations have confirmed the aromaticity of the heteroaromatic system and determined its electronic structure that is relevant to applications in materials and supramolecular chemistry.
Co-reporter:Ling Yuan, Bobby G. Sumpter, Khalil A. Abboud and Ronald K. Castellano
New Journal of Chemistry 2008 vol. 32(Issue 11) pp:1924-1934
Publication Date(Web):16 Jul 2008
DOI:10.1039/B808818G
3,5-Disubstituted piperidones provide an opportunity to explore donor–acceptor through-bond interactions in the context of molecular and supramolecular structure. The crystal structure of cis-3,5-dibenzyl-1-phenylpiperidin-4-one 3 is disordered and the lattice accommodates a ∼3 : 1 ratio of the N-Ph equatorial (3-eq) and N-Ph axial (3-ax) epimers, based on refined values of occupancy factors. The fortuitous result allows a side-by-side comparison of the two configurations with respect to their donor–acceptor through-bond interactions. The energy difference between 3-ax and 3-eq (ΔEax–eq) has been evaluated in the gas phase using extensive first principles calculations, and for many levels of theory this difference parallels the experimentally-observed configurational ratio in the solid state (where the epimers share nearly identical packing environments). The calculations further show a difference in the through-bond stabilization for 3-ax and 3-eq, with larger contributions for 3-ax. Natural bond order (NBO) analysis quantifies the delocalization of the donor nitrogen lone pair into the adjacent carbon–carbon bonds and carbonyl acceptor for the 3-axepimer. The work concludes that molecular-level structural perturbations that arise from or otherwise influence through-bond donor–acceptor interactions have consequences on solid-state and supramolecular assembly structure.
Co-reporter:AndrewJ. Lampkins;Yan Li;Alexre AlAbbas;KhalilA. Abboud Dr.;Ion Ghiviriga Dr. ;RonaldK. Castellano
Chemistry - A European Journal 2008 Volume 14( Issue 5) pp:1452-1463
Publication Date(Web):
DOI:10.1002/chem.200701220
Abstract
Reported are the syntheses of ester-functionalized (6–8) and alkyl-substituted (9) 1-aza-adamantanones; the easy handling of the compounds provides an opportunity to comprehensively study the fundamental changes in structure and reactivity that can accompany the donor–acceptor arrangement in rigid β-aminoketones. X-ray structural analysis of trione 6 and dione 7 reveals bond length and angle variations consistent with through-bond (hyperconjugative) donor–acceptor interactions. Observed is a shortening of the CN bond, elongation of the central CC bond (to ≈1.6 Å), and a significant pyramidalization of the carbonyl carbon within the donor–σ-acceptor pathway. UV/Vis spectra of 6–9 show a new absorption maximum (λmax=260–275 nm in three solvents), the so-called “σ-coupled transition”; the molar absorptivity scales with the number of carbonyl groups (for trione 6, ε ≈3000, for dione 7, ε ≈2000) and the band reversibly disappears upon addition of acid. IR and 13C NMR spectroscopic data show trends consistent with through-bond donation to the carbonyl acceptor groups and commensurate weakening of the carbonyl π bond. High yielding acid-mediated fragmentations are used to illustrate the effects of the donor–acceptor arrangement on the reactivity of the molecules. Given that donor–σ-acceptor molecules have recently been found to show self-assembly behavior and macromolecular properties linked to their unusual structure, the current analysis encourages further consideration of the systems in advanced materials applications.
Co-reporter:Roslyn S. Butler, Andrea K. Myers, Prabhu Bellarmine, Khalil A. Abboud and Ronald K. Castellano
Journal of Materials Chemistry A 2007 vol. 17(Issue 19) pp:1863-1865
Publication Date(Web):26 Feb 2007
DOI:10.1039/B618171F
Donor–acceptor purines have been prepared that show near unity fluorescence quantum yields in organic solution, a step toward using simple biorelevant heterocycles in optoelectronic and photonic applications.
Co-reporter:Alisha M. Martin, Roslyn S. Butler, Ion Ghiviriga, Rachel E. Giessert, Khalil A. Abboud and Ronald K. Castellano
Chemical Communications 2006 (Issue 42) pp:4413-4415
Publication Date(Web):07 Sep 2006
DOI:10.1039/B610239E
The first discrete, self-complementary, quadruply hydrogen-bonded complexes based on the 2,6-diaminopurine (DAP) scaffold have been prepared; regioselective urea formation at the C(2) amino group of the heterocycle allows intermolecular dimerization (Kdim
∼ 1–1.6 × 103 M−1 in CDCl3) through a DADA hydrogen bonding motif.
Co-reporter:Emmanuel A. Meyer Dipl.-Chem. Dr.;François Diederich Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 11) pp:
Publication Date(Web):13 MAR 2003
DOI:10.1002/anie.200390319
Intermolecular interactions involving aromatic rings are key processes in both chemical and biological recognition. Their understanding is essential for rational drug design and lead optimization in medicinal chemistry. Different approaches—biological studies, molecular recognition studies with artificial receptors, crystallographic database mining, gas-phase studies, and theoretical calculations—are pursued to generate a profound understanding of the structural and energetic parameters of individual recognition modes involving aromatic rings. This review attempts to combine and summarize the knowledge gained from these investigations. The review focuses mainly on examples with biological relevance since one of its aims it to enhance the knowledge of molecular recognition forces that is essential for drug development.
Co-reporter:Emmanuel A. Meyer Dipl.-Chem. Dr.;François Diederich Dr.
Angewandte Chemie 2003 Volume 115(Issue 11) pp:
Publication Date(Web):13 MAR 2003
DOI:10.1002/ange.200390290
Zwischenmolekulare Wechselwirkungen unter Beteiligung aromatischer Ringe sind Schlüsselvorgänge sowohl in chemischen als auch in biologischen Erkennungsprozessen. Ihr Verständnis ist für das rationale Wirkstoffdesign und für die Leitstrukturoptimierung in der Medizinischen Chemie essenziell. Unterschiedliche Ansätze werden für ein tiefergehendes Verständnis der strukturellen und energetischen Parameter einzelner Erkennungsarten mit aromatischen Substraten verfolgt: erwähnt seien biologische Untersuchungen, Studien der molekularen Erkennung mit künstlichen Rezeptoren, Suchen in kristallographischen Datenbanken, Gasphasenstudien und theoretische Untersuchungen. Dieser Aufsatz versucht, diese Wissensgebiete zu vereinen und die aus zahlreichen Untersuchungen gewonnenen Erkenntnisse zusammenzufassen. Er widmet sich hauptsächlich Beispielen mit biologischer Relevanz mit dem Ziel, die für die Wirkstoffentwicklung wichtigen Kenntnisse der molekularen Erkennung zu vertiefen.
Co-reporter:Emmanuel A. Meyer Dipl.-Chem. Dr.;François Diederich Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 35) pp:
Publication Date(Web):15 SEP 2003
DOI:10.1002/anie.200390574
Co-reporter:Emmanuel A. Meyer Dipl.-Chem. Dr.;François Diederich Dr.
Angewandte Chemie 2003 Volume 115(Issue 35) pp:
Publication Date(Web):15 SEP 2003
DOI:10.1002/ange.200390601
Co-reporter:Yixing Yang, Pamela Cohn, Sang-Hyun Eom, Khalil A. Abboud, Ronald K. Castellano and Jiangeng Xue
Journal of Materials Chemistry A 2013 - vol. 1(Issue 16) pp:NaN2874-2874
Publication Date(Web):2013/02/18
DOI:10.1039/C3TC00734K
We report efficient ultraviolet (UV)-violet organic light-emitting devices (OLEDs) based on highly fluorescent donor–acceptor purine molecules, which can generate tunable emission from 350 nm to 450 nm in solution by using different electron donor and acceptor arrangements on the heterocycles as reported previously. Here, external quantum efficiencies (EQEs) up to ηEQE = 1.6% are achieved for the multilayer OLEDs based on purine 2, with UV emission peaked at 393 nm, as compared to previously reported purine 1 based OLEDs with ηEQE = 3.1% and peak emission at 433 nm. The efficiencies of the OLEDs based on the two purine molecules are among the highest reported to date with emission peak wavelengths below 450 nm. By using a range of charge transport and host materials, we show that appropriate energy level alignment in multilayer OLED devices is imperative to achieve UV emission and prevent undesired emission from other layers or interfaces.
Co-reporter:Matthew B. Baker, Ion Ghiviriga and Ronald K. Castellano
Chemical Science (2010-Present) 2012 - vol. 3(Issue 4) pp:NaN1099-1099
Publication Date(Web):2011/12/20
DOI:10.1039/C2SC00943A
Synthetic access to discrete multifunctionalized molecules remains a challenge but is critical for continued advances in materials science, catalysis, and chemical biology. Here, the aminolysis of electronically coupled lactones is introduced as an approach to achieve molecular multifunctionalization efficiently, under mild conditions, and without protecting groups or by-products. The one-pot sequential aminolysis of benzotrifuranone, a C3h-symmetric trilactone, to produce a trifunctionalized target in ∼85% yield over one day is highlighted.
Co-reporter:Benjamin M. Schulze, Davita L. Watkins, Jing Zhang, Ion Ghiviriga and Ronald K. Castellano
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 40) pp:NaN7936-7936
Publication Date(Web):2014/08/28
DOI:10.1039/C4OB01373E
Reported is characterization of the self-assembly of π-conjugated oligomers, molecules studied recently in photovoltaic devices, using variable temperature diffusion ordered spectroscopy (VT-DOSY). Iterative fitting of diffusion coefficient versus temperature data to a modified Stokes–Einstein equation, molecular modelling, and comparison to non-assembling model compounds, has allowed estimation of assembly size, shape, and molecularity.
Co-reporter:Benjamin M. Schulze, Nathan T. Shewmon, Jing Zhang, Davita L. Watkins, John P. Mudrick, Weiran Cao, Raghida Bou Zerdan, Anthony J. Quartararo, Ion Ghiviriga, Jiangeng Xue and Ronald K. Castellano
Journal of Materials Chemistry A 2014 - vol. 2(Issue 5) pp:NaN1549-1549
Publication Date(Web):2013/12/04
DOI:10.1039/C3TA13529B
Reported is a systematic molecular structure–property relationship study to evaluate the consequences of dedicated H-bonding interactions between molecular electron donors on molecular assembly, absorption, charge collection, and performance in small-molecule bulk heterojunction organic photovoltaic devices. Three families of branched quaterthiophene donor chromophores have been synthesized with members that share nearly identical electronic and optical properties in the molecularly dispersed state but are either capable or incapable of self-association by hydrogen bonding (H-bonding). Phthalhydrazide-functionalized quaterthiophenes are H-bond “active” and show signatures of H-bond promoted assembly in solution (by 1H NMR) and in both neat and blended (with C60) films (by IR). Compared to control compounds with H-bonding “turned off”, the H-bonded derivatives show red-shifted thin film absorption (neat and as blends with C60), different colors as bulk solids, and increased decomposition and melt temperatures. Photovoltaic devices made from blends of H-bonded donor molecules with C60 as the electron acceptor show improved charge collection length and external quantum efficiency resulting in a more than two-fold enhancement in power conversion efficiency relative to non-H-bonding controls, from 0.49% to 1.04%. We anticipate this approach could be generalized to include other donor chromophores with lower optical gap to harvest more longer-wavelength photons and achieve higher power conversion efficiencies.
Co-reporter:Roslyn S. Butler, Andrea K. Myers, Prabhu Bellarmine, Khalil A. Abboud and Ronald K. Castellano
Journal of Materials Chemistry A 2007 - vol. 17(Issue 19) pp:NaN1865-1865
Publication Date(Web):2007/02/26
DOI:10.1039/B618171F
Donor–acceptor purines have been prepared that show near unity fluorescence quantum yields in organic solution, a step toward using simple biorelevant heterocycles in optoelectronic and photonic applications.

![2,5-bis(2-(2-(2-methoxyethoxy)ethoxy)ethyl)-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione](http://img.cochemist.com/ccimg/1296200/1296131-04-8.png)
![2,5-bis(2-(2-(2-methoxyethoxy)ethoxy)ethyl)-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione](http://img.cochemist.com/ccimg/1296200/1296131-04-8_b.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 2,5-bis(2-ethylhexyl)-3,6-bis(5''-hexyl[2,2':5',2''-terthiophen]-5-yl)-2,5-dihydro-](/data/chemimg/347100/1093468-95-1.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 2,5-bis(2-ethylhexyl)-3,6-bis(5''-hexyl[2,2':5',2''-terthiophen]-5-yl)-2,5-dihydro-](/data/chemimg/347100/1093468-95-1_b.png)






![Tricyclo[8.2.2.24,7]hexadeca-4,6,10,12,13,15-hexaene,5-bromo-](http://img.cochemist.com/ccimg/2000/1908-61-8.png)
![Tricyclo[8.2.2.24,7]hexadeca-4,6,10,12,13,15-hexaene,5-bromo-](http://img.cochemist.com/ccimg/2000/1908-61-8_b.png)
![2,5-Bis(2-ethylhexyl)-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione](http://img.cochemist.com/ccimg/1185900/1185885-86-2.png)
![2,5-Bis(2-ethylhexyl)-3,6-di(thiophen-2-yl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione](http://img.cochemist.com/ccimg/1185900/1185885-86-2_b.png)




![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3,6-bis(5-bromo-2-thienyl)-2,5-bis(2-ethylhexyl)-2,5-dihydro-](/data/chemimg/139400/1000623-95-9.png)
![Pyrrolo[3,4-c]pyrrole-1,4-dione, 3,6-bis(5-bromo-2-thienyl)-2,5-bis(2-ethylhexyl)-2,5-dihydro-](/data/chemimg/139400/1000623-95-9_b.png)












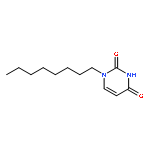
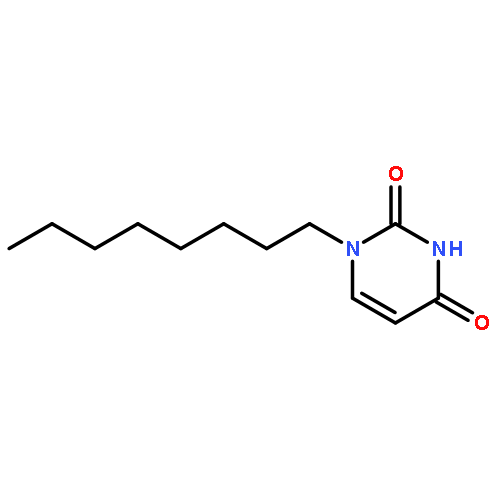
![Ethanol, 2-[2-(2-methoxyethoxy)ethoxy]-, 1-(4-methylbenzenesulfonate)](/data/chemimg/636900/62921-74-8.png)
![Ethanol, 2-[2-(2-methoxyethoxy)ethoxy]-, 1-(4-methylbenzenesulfonate)](/data/chemimg/636900/62921-74-8_b.png)