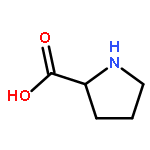
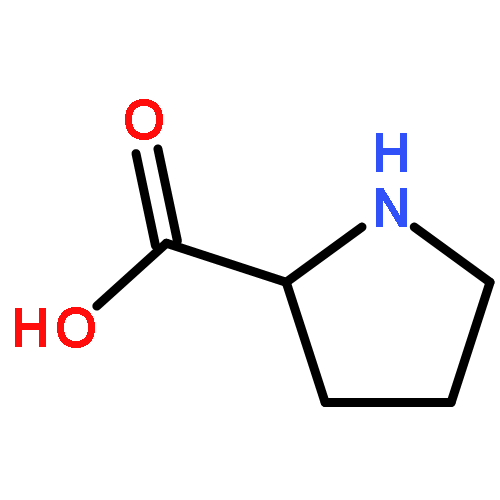
Co-reporter:Weitong Ren, Wenfei Li, Jun Wang, Jian Zhang, and Wei Wang
The Journal of Physical Chemistry B October 26, 2017 Volume 121(Issue 42) pp:9799-9799
Publication Date(Web):September 25, 2017
DOI:10.1021/acs.jpcb.7b06919
Allosteric proteins are featured by energetic degeneracy of two (or more) functionally relevant conformations, therefore their energy landscapes are often locally frustrated. How such frustration affects the protein folding/binding dynamics is not well understood. Here, by using molecular simulations we study the consequences of local frustration in the dimerization dynamics of allosteric proteins based on a homodimer protein S100A12. Despite of the structural symmetry of the two EF-hand motifs in the three-dimensional structures, the S100A12 homodimer shows allosteric behaviors and local frustration only in half of its structural elements, i.e., the C-terminal EF-hand. We showed that such spatially asymmetric location of frustration leads to asymmetric dimerization pathways, in which the dimerization is dominantly initiated by the interchain binding of the minimally frustrated N-terminal EF-hands, achieving optimal balance between the requirements of rapid conformational switching and interchain assembling to the energy landscapes. We also showed that the local frustration, as represented by the double-basin topography of the energy landscape, gives rise to multiple cross-linked dimerization pathways, in which the dimerization is coupled with the allosteric motions of the C-terminal EF-hands. Binding of metal ions tends to reshape the energy landscape and modulate the dimerization pathways. In addition, by employing the frustratometer method, we showed that the highly frustrated residue-pairs in the C-terminal EF-hand are partially unfolded during the conformational transitions of the native homodimer, leading to lowing of free energy barrier. Our results revealed tight interplay between the local frustration of the energy landscape and the dimerization dynamics for allosteric proteins.
Co-reporter:Cheng Tan, Wenfei Li, and Wei Wang
The Journal of Physical Chemistry B 2013 Volume 117(Issue 50) pp:15917-15925
Publication Date(Web):November 22, 2013
DOI:10.1021/jp4052165
Protein TFIIIA is composed of nine tandemly arranged Cys2His2 zinc fingers. It can bind either to the 5S RNA gene as a transcription factor or to the 5S RNA transcript as a chaperone. Although structural and biochemical data provided valuable information on the recognition between the TFIIIIA and the 5S DNA/RNA, the involved conformational motions and energetic factors contributing to the binding affinity and specificity remain unclear. In this work, we conducted MD simulations and MM/GBSA calculations to investigate the binding-induced conformational changes in the recognition of the 5S RNA by the central three zinc fingers of TFIIIA and the energetic factors that influence the binding affinity and specificity at an atomistic level. Our results revealed drastic interdomain conformational changes between these three zinc fingers, involving the exposure/burial of several crucial DNA/RNA binding residues, which can be related to the competition between DNA and RNA for the binding of TFIIIA. We also showed that the specific recognition between finger 4/finger 6 and the 5S RNA introduces frustrations to the nonspecific interactions between finger 5 and the 5S RNA, which may be important to achieve optimal binding affinity and specificity.
Co-reporter:Wenhui Xi, Wenfei Li, and Wei Wang
The Journal of Physical Chemistry B 2012 Volume 116(Issue 25) pp:7398-7405
Publication Date(Web):June 6, 2012
DOI:10.1021/jp300389g
Population of aggregation-prone conformers for the monomeric amyloid-β (Aβ) can dramatically speed up its fibrillar aggregation. In this work, we study the effect of preformed template on the conformational distributions of the monomeric Aβ by replica exchange molecular dynamics. Our results show that the template consisting of Aβ peptides with cross-β structure can induce the formation of β-rich conformations for the monomeric Aβ, which is the key feature of the aggregation-prone conformers. Similar effect is observed when the hIAPP peptides and poly alanine peptides were used as templates, suggesting that the template effect is insensitive to the sequence details of the template peptides. In comparison, the template with helical structure has no significant effects on the β-propensity of the monomeric Aβ. Analysis to the interaction details revealed that the template tends to disrupt the intrapeptide interactions of the monomeric Aβ, which are absent in the fibrillar state, suggesting that the preformed template can reorganize the intrapeptide interactions of the monomeric Aβ during the capturing stage and reduce the energy frustrations for the fibrillar aggregations.
Co-reporter:Guanghong Zuo, Wenfei Li, Jian Zhang, Jin Wang and Wei Wang
The Journal of Physical Chemistry B 2010 Volume 114(Issue 17) pp:5835-5839
Publication Date(Web):April 14, 2010
DOI:10.1021/jp904573r
The folding of a small RNA tetraloop hairpin is studied based on intensive molecular dynamics simulation, aiming to understand the folding mechanism of this small and fast RNA folder. Our results showed that this RNA hairpin has very complicated folding behavior in spite of its small size. It is found that the folding transition has low cooperativity. Instead of a two-state folding, four major states are observed, including the native state, the intermediate, the unfolded state, and the misfolded state. The misfolded state is mainly stabilized by the non-native hydrogen bonds, and is more compact. Two potential folding pathways, in which two basepairs formed with different order, are observed, and the pathway with the inboard basepair formed before the terminal one is much more favorable, and dominates the folding of the RNA hairpin.

![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)