Co-reporter:Uyen B. Chu, Sevahn K. Vorperian, Kenneth Satyshur, Kelsey Eickstaedt, Nicholas V. Cozzi, Timur Mavlyutov, Abdol R. Hajipour, and Arnold E. Ruoho
Biochemistry 2014 Volume 53(Issue 18) pp:
Publication Date(Web):April 14, 2014
DOI:10.1021/bi500175p
Indolethylamine-N-methyltransferase (INMT) is a Class 1 transmethylation enzyme known for its production of N,N-dimethyltryptamine (DMT), a hallucinogen with affinity for various serotonergic, adrenergic, histaminergic, dopaminergic, and sigma-1 receptors. DMT is produced via the action of INMT on the endogenous substrates tryptamine and S-adenosyl-l-methionine (SAM). The biological, biochemical, and selective small molecule regulation of INMT enzyme activity remain largely unknown. Kinetic mechanisms for inhibition of rabbit lung INMT (rabINMT) by the product, DMT, and by a new novel tryptamine derivative were determined. After Michaelis–Menten and Lineweaver–Burk analyses had been applied to study inhibition, DMT was found to be a mixed competitive and noncompetitive inhibitor when measured against tryptamine. The novel tryptamine derivative, N-[2-(1H-indol-3-yl)ethyl]-N′,N′-dimethylpropane-1,3-diamine (propyl dimethyl amino tryptamine or PDAT), was shown to inhibit rabINMT by a pure noncompetitive mechanism when measured against tryptamine with a Ki of 84 μM. No inhibition by PDAT was observed at 2 mM when it was tested against structurally similar Class 1 methyltransferases, such as human phenylethanolamine-N-methyltransferase (hPNMT) and human nicotinamide-N-methyltransferase (hNNMT), indicating selectivity for INMT. The demonstration of noncompetitive mechanisms for INMT inhibition implies the presence of an inhibitory allosteric site. In silico analyses using the computer modeling software Autodock and the rabINMT sequence threaded onto the human INMT (hINMT) structure (Protein Data Bank entry 2A14) identified an N-terminal helix–loop–helix non-active site binding region of the enzyme. The energies for binding of DMT and PDAT to this region of rabINMT, as determined by Autodock, were −6.34 and −7.58 kcal/mol, respectively. Assessment of the allosteric control of INMT may illuminate new biochemical pathway(s) underlying the biology of INMT.
Co-reporter:Uyen B. Chu, Subramaniam Ramachandran, Abdol R. Hajipour, and Arnold E. Ruoho
Biochemistry 2013 Volume 52(Issue 5) pp:
Publication Date(Web):January 16, 2013
DOI:10.1021/bi301517u
The sigma-1 receptor is a ligand-regulated endoplasmic reticulum (ER) resident chaperone involved in the maintenance of cellular homeostasis. Coupling of the sigma-1 receptor with various ER and/or plasma membrane ion channels is associated with its ability to regulate the locomotor activity and cellular proliferation produced in response to sigma-1 receptor ligands. A number of endogenous small molecules bind to the sigma-1 receptor and have been shown to regulate its activity; these include progesterone, N,N-dimethyltryptamine, d-erythro-sphingosine, and/or other endogenous lipids. We previously reported the synthesis of long chain N-alkylamine derivatives and the characterization of the structure–activity relationship between the chain length of N-alkylamine and affinities at the sigma-1 receptor. Here, we present data demonstrating the photoincorporation of one of these N-alkylamine derivatives, N-[3-(4-nitrophenyl)propyl]-N-dodecylamine (4-NPPC12), to the sigma-1 receptor. Matrix-assisted laser desorption ionization time-of-flight and tandem mass spectrometry showed that 4-NPPC12 photoinserted at histidine 154 of the derivatized population of the sigma-1 receptor. Interestingly, light-dependent photoinsertion of 4-NPPC12 resulted in an enhanced electrophoretic mobility of only 50% of the derivatized receptor molecules as assessed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The proposed binding and reactivity of 4-NPPC12 evoke a ligand binding model for the sigma-1 receptor that likely involves a receptor dimer and/or oligomer.
Co-reporter:Dr. Lian-Wang Guo; Abdol R. Hajipour;Dr. Kerim Karaoglu;Dr. Timur A. Mavlyutov; Arnold E. Ruoho
ChemBioChem 2012 Volume 13( Issue 15) pp:2277-2289
Publication Date(Web):
DOI:10.1002/cbic.201200427
Abstract
Sigma (σ) receptors are unique non-opioid binding sites that are associated with a broad range of disease states. Sigma-2 receptors provide a promising target for diagnostic imaging and pharmacological interventions to curb tumor progression. Most recently, the progesterone receptor (PGRMC1, 25 kDa) has been shown to have σ2 receptor-like binding properties, thus highlighting the need to understand the biological function of an 18 kDa protein that exhibits σ2-like photoaffinity labeling (denoted here as σ2-18k) but the amino acid sequence of which is not known. In order to provide new tools for the study of the σ2-18k protein, we have developed bifunctional σ receptor ligands each bearing a benzophenone photo-crosslinking moiety and an alkyne group to which an azide-containing biotin affinity tag can be covalently attached through click chemistry after photo-crosslinking. Although several compounds showed favorable σ2 binding properties, the highest affinity (2 nM) and the greatest potency in blocking photolabeling of σ2-18k by a radioactive photoaffinity ligand was shown by compound 22. These benzophenone-alkyne σ receptor ligands might therefore be amenable for studying the σ2-18k protein through chemical biology approaches. To the best of our knowledge, these compounds represent the first reported benzophenone-containing clickable σ receptor ligands, which might potentially have broad applications based on the “plugging in” of various tags.
Co-reporter:Uyen B. Chu, Abdol R. Hajipour, Subramaniam Ramachandran, and Arnold E. Ruoho
Biochemistry 2011 Volume 50(Issue 35) pp:
Publication Date(Web):July 26, 2011
DOI:10.1021/bi2004872
Sigma receptors are small membrane proteins implicated in a number of pathophysiological conditions, including drug addiction, psychosis, and cancer; thus, small molecule inhibitors of sigma receptors have been proposed as potential pharmacotherapeutics for these diseases. We previously discovered that endogenous monochain N-alkyl sphingolipids, including d-erythro-sphingosine, sphinganine, and N,N-dimethylsphingosine, bind to the sigma-1 receptor at physiologically relevant concentrations [Ramachandran, S., et al. (2009) Eur. J. Pharmacol. 609, 19–26]. Here, we investigated several N-alkylamines of varying chain lengths as sigma receptor ligands. Although the KI values for N-alkylamines were found to be in the micromolar range, when N-3-phenylpropyl and N-3-(4-nitrophenyl)propyl derivatives of butylamine (1a and 1b, respectively), heptylamine (2a and 2b, respectively), dodecylamine (3a and 3b, respectively), and octadecylamine (4a and 4b, respectively) were evaluated as sigma receptor ligands, we found that these compounds exhibited nanomolar affinities with both sigma-1 and sigma-2 receptors. A screen of high-affinity ligands 2a, 2b, 3a, and 3b against a variety of other receptors and/or transporters confirmed these four compounds to be highly selective mixed sigma-1 and sigma-2 ligands. Additionally, in HEK-293 cells reconstituted with Kv1.4 potassium channel and the sigma-1 receptor, these derivatives were able to inhibit the outward current from the channel, consistent with sigma receptor modulation. Finally, cytotoxicity assays showed that 2a, 2b, 3a, and 3b were highly potent against a number of cancer cell lines, demonstrating their potential utility as mixed sigma-1 and sigma-2 receptor anticancer agents.
Co-reporter:Abdol R. Hajipour, Lian-Wang Guo, Arindam Pal, Timur Mavlyutov, Arnold E. Ruoho
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 24) pp:7435-7440
Publication Date(Web):15 December 2011
DOI:10.1016/j.bmc.2011.10.046
The sigma-2 (σ2) receptor has been suggested to be a promising target for pharmacological interventions to curb tumor progression. Development of σ2-specific ligands, however, has been hindered by lack of understanding of molecular determinants that underlie selective ligand-σ2 interactions. Here we have explored effects of electron donating and withdrawing groups on ligand selectivity for the σ2 versus σ1 receptor using new benzamide-isoquinoline derivatives. The electron-donating methoxy group increased but the electron-withdrawing nitro group decreased σ2 affinity. In particular, an extra methoxy added to the para-position (5e) of the benzamide phenyl ring of 5f dramatically improved (631 fold) the σ2 selectivity relative to the σ1 receptor. This para-position provided a sensitive site for effective manipulation of the sigma receptor subtype selectivity using either the methoxy or nitro substituent. Our study provides a useful guide for further improving the σ2-over-σ1 selectivity of new ligands.
Co-reporter:Abdol R. Hajipour, Dominique Fontanilla, Uyen B. Chu, Marty Arbabian, Arnold E. Ruoho
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 12) pp:4397-4404
Publication Date(Web):15 June 2010
DOI:10.1016/j.bmc.2010.04.078
The sigma-1 receptor is a unique non-opioid, non-PCP binding site that has been implicated in many different pathophysiological conditions including psychosis, drug addiction, retinal degeneration and cancer. Based on the structure of fenpropimorph, a high affinity (Ki = 0.005 nM)1 sigma-1 receptor ligand and strong inhibitor of the yeast sterol isomerase (ERG2), we previously deduced a basic sigma-1 receptor pharmacophore or chemical backbone composed of a phenyl ring attached to a di-substituted nitrogen atom via an alkyl chain.2 Here, we report the design and synthesis of various N,N-dialkyl or N-alkyl-N-aralkyl derivatives based on this pharmacophore as well as their binding affinities to the sigma-1 receptor. We introduce three high affinity sigma-1 receptor compounds, N,N-dibutyl-3-(4-fluorophenyl)propylamine (9), N,N-dibutyl-3-(4-nitrophenyl)propylamine (3), and N-propyl-N′-4-aminophenylethyl-3-(4-nitrophenyl)propylamine (20) with Ki values of 17.7 nM, 0.36 nM, and 6 nM, respectively. In addition to sigma receptor affinity, we show through cytotoxicity assays that growth inhibition of various tumor cell lines occurs with our high affinity N,N-dialkyl or N-alkyl-N-aralkyl derivatives.
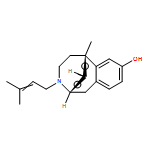
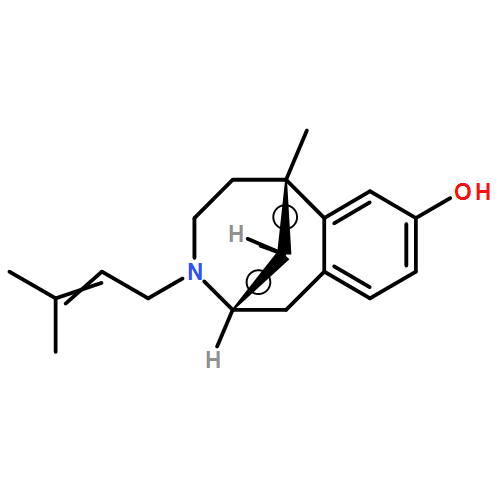
Co-reporter:Dominique Fontanilla;Molly Johannessen;Abdol R. Hajipour;Nicholas V. Cozzi;Meyer B. Jackson;Arnold E. Ruoho
Science 2009 Vol 323(5916) pp:934-937
Publication Date(Web):13 Feb 2009
DOI:10.1126/science.1166127
Abstract
The sigma-1 receptor is widely distributed in the central nervous system and periphery. Originally mischaracterized as an opioid receptor, the sigma-1 receptor binds a vast number of synthetic compounds but does not bind opioid peptides; it is currently considered an orphan receptor. The sigma-1 receptor pharmacophore includes an alkylamine core, also found in the endogenous compound N,N-dimethyltryptamine (DMT). DMT acts as a hallucinogen, but its receptor target has been unclear. DMT bound to sigma-1 receptors and inhibited voltage-gated sodium ion (Na+) channels in both native cardiac myocytes and heterologous cells that express sigma-1 receptors. DMT induced hypermobility in wild-type mice but not in sigma-1 receptor knockout mice. These biochemical, physiological, and behavioral experiments indicate that DMT is an endogenous agonist for the sigma-1 receptor.
Co-reporter:Dominique Fontanilla, Abdol R. Hajipour, Arindam Pal, Uyen B. Chu, Marty Arbabian and Arnold E. Ruoho
Biochemistry 2008 Volume 47(Issue 27) pp:
Publication Date(Web):June 12, 2008
DOI:10.1021/bi800564j
Radioiodinated photoactivatable photoprobes can provide valuable insights regarding protein structure. Previous work in our laboratory showed that the cocaine derivative and photoprobe 3-[125I]iodo-4-azidococaine ([125I]IACoc) binds to the sigma-1 receptor with 2−3 orders of magnitude higher affinity than cocaine [Kahoun, J. R. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 1393−1397]. Using this photoprobe, we demonstrated the insertion site for [125I]IACoc to be Asp188 [Chen, Y. (2007) Biochemistry 46, 3532−3542], which resides in the proposed steroid binding domain-like II (SBDLII) region (amino acids 176−194) [Pal, A. (2007) Mol. Pharmacol. 72, 921−933]. An additional photoprobe based on the sigma-1 receptor ligand fenpropimorph, 1-N-(2-3-[125I]iodophenyl)propane ([125I]IAF), was found to label a peptide in both the SBDLII and steroid binding domain-like I (SBDLI) (amino acids 91−109) [Pal, A. (2007) Mol. Pharmacol. 72, 921−933]. In this report, we describe two novel strategically positioned carrier-free, radioiodinated photoaffinity labels specifically designed to probe the putative “nitrogen interacting region” of sigma-1 receptor ligands. These two novel photoprobes are (−)-methyl 3-(benzoyloxy)-8-2-(4-azido-3-[125I]iodobenzene)-1-ethyl-8-azabicyclo[3.2.1]octane-2-carboxylate ([125I]-N-IACoc) and N-propyl-N-(4-azido-3-iodophenylethyl)-3-(4-fluorophenyl)propylamine ([125I]IAC44). In addition to reporting their binding affinities to the sigma-1 and sigma-2 receptors, we show that both photoaffinity labels specifically and covalently derivatized the pure guinea pig sigma-1 receptor (26.1 kDa) [Ramachandran, S. (2007) Protein Expression Purif. 51, 283−292]. Cleavage of the photolabeled sigma-1 receptor using Endo Lys C and cyanogen bromide (CNBr) revealed that the [125I]-N-IACoc label was located primarily in the N-terminus and SBDLI-containing peptides of the sigma-1 receptor, while [125I]IAC44 was found in peptide fragments consistent with labeling of both SBDLI and SBDLII.
Co-reporter:Jikui Song;Lian-Wang Guo;Hakim Muradov;Nikolai O. Artemyev;Arnold E. Ruoho;John L. Markley;
Proceedings of the National Academy of Sciences 2008 105(5) pp:1505-1510
Publication Date(Web):January 29, 2008
DOI:10.1073/pnas.0709558105
The retinal phosphodiesterase (PDE6) inhibitory γ-subunit (PDEγ) plays a central role in vertebrate phototransduction through
alternate interactions with the catalytic αβ-subunits of PDE6 and the α-subunit of transducin (αt). Detailed structural analysis of PDEγ has been hampered by its intrinsic disorder. We present here the NMR solution structure
of PDEγ, which reveals a loose fold with transient structural features resembling those seen previously in the x-ray structure
of PDEγ46–87 when bound to αt in the transition-state complex. NMR mapping of the interaction between PDEγ46–87 and the chimeric PDE5/6 catalytic domain confirmed that C-terminal residues 74–87 of PDEγ are involved in the association
and demonstrated that its W70 indole group, which is critical for subsequent binding to αt, is left free at this stage. These results indicate that the interaction between PDEγ and αt during the phototransduction cascade involves the selection of preconfigured transient conformations.
Co-reporter:Timur A Mavlyutov;Arnold E Ruoho
Journal of Molecular Signaling 2007 Volume 2( Issue 1) pp:
Publication Date(Web):2007 December
DOI:10.1186/1750-2187-2-8
Sigma-1 receptors are involved in regulation of neuronal activities presumably through regulation of the activity of ion channels. Sigma-1 receptors also play a role in growth and metastasis of cancerous cells. Intracellular distribution of sigma-1 receptors have been linked to sphingolipid-enriched domains.We report that in CHO-K1 cells sigma-1 receptors target to focal adhesion contacts (FAC) where they colocalize with Talin and Kv1.4 potassium channels. The levels of sigma-1 receptors in the FAC were significantly increased by application of sigma-1 receptor ligands and by filamentous actin (F-actin) polymerization with phalloidin. The total length of FAC (measured by the focal adhesion marker, talin) was concomitantly increased in the presence of sigma-1 receptors upon phalloidin treatment. Only sigma-1 receptor ligands, however, resulted in an increase of sigma-1 receptors in the FAC, independent of talin. Additionally, a novel approach was utilized to allow an assessment of the half life of endogenous sigma-1 receptors in CHO-K1 cells, which was measured to be at least 72 hours.Ligand activated sigma-1 receptors translocate into FAC from a pool of receptors stored in ER lipid rafts presumably for inhibition of Kv1.4 channels. Stabilization of actin filaments is likely to be important for targeting sigma-1 receptors to Focal Adhesion Contacts in CHO-K1 cells.
Co-reporter:Timur A. Mavlyutov, Lian-Wang Guo, Miles L. Epstein, Arnold E. Ruoho
Journal of Pharmacological Sciences (January 2015) Volume 127(Issue 1) pp:10-16
Publication Date(Web):1 January 2015
DOI:10.1016/j.jphs.2014.12.013
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disease affecting spinal cord motoneurons (MN) with an associative connection to Frontotemporal Lobar Dementia (FTLD). The endoplasmic reticulum (ER) bound Sigma-1 Receptor (S1R) chaperone protein localizes to specialized ER cisternae within 10 nm of the plasma membrane in spinal cord ventral horn cholinergic post synaptic C-terminals. Removal of the S1R gene in the Superoxide Dismutase-1 (SOD-1) mouse model of ALS exacerbated the neurodegenerative condition and resulted in a significantly reduced longevity when compared to the SOD-1/S1R wild type (WT) mouse. The proposed amelioration of the ALS phenotype by the S1R is likely due to a “brake” on excitation of the MN as evidenced by a reduction in action potential generation in the MN of the WT when compared to the S1R KO mouse MN. Although the precise signal transduction pathway(s) regulated by the S1R in the MN has/have not been elucidated at present, it is likely that direct or indirect functional interactions occur between the S1R in the ER cisternae with voltage gated potassium channels and/or with muscarinic M2 receptor signaling in the post synaptic plasma membrane. Possible mechanisms for regulation of MN excitability by S1R are discussed.
Co-reporter:Subramaniam Ramachandran, Hongliang Lu, Usha Prabhu, Arnold E. Ruoho
Protein Expression and Purification (February 2007) Volume 51(Issue 2) pp:283-292
Publication Date(Web):1 February 2007
DOI:10.1016/j.pep.2006.07.019
Sigma receptors once considered as a class of opioid receptors are now regarded as unique orphan receptors, distinguished by the ability to bind various pharmacological agents such as the progesterone (steroid), haloperidol (anti-psychotic), and drugs of abuse such as cocaine and methamphetamine. The sigma-1 receptor is a 223 amino acid protein, proposed to have two transmembrane segments. We have developed a scheme for the purification of the guinea pig sigma-1 receptor following overexpression in Escherichia coli as a maltose binding protein (MBP) fusion and extraction with Triton X-100. Affinity chromatography using an amylose column and Ni2+ affinity column was used to purify the sigma-1 receptor. The sigma-1 receptor purified by this method is a 26 kDa polypeptide as assessed by SDS–PAGE, binds sigma ligands with high affinity and can be specifically photoaffinity labeled with the sigma-1 receptor photoprobe, [125I]-iodoazidococaine. Ligand binding using [3H]-(+)-pentazocine indicated that approximately half of the purified protein in Triton X-100 bound to radioligand. The MBP-sigma-1 receptor and the sigma-1 receptor in 0.5% triton were maximally stable for approximately two weeks at −20 °C in buffer containing 30% glycerol.
Co-reporter:B. Torres, A.E. Ruoho
Neuroscience (14 February 2014) Volume 259() pp:194-202
Publication Date(Web):14 February 2014
DOI:10.1016/j.neuroscience.2013.11.059
•The VMAT2 N-terminus enables sequestration.•A GST-construct of the N-terminus undergoes phosphorylation by PKC at serines 15 and/or 18.•The charged state of N-terminus serines 15 and 18 regulates sequestration by the VMAT2.•The charged state of N-terminus serines 15 and 18 regulates the VMAT2 efflux response to methamphetamine.The 20 amino acid (AA) N-terminus of the vesicular monoamine transporter 2 (VMAT2) was examined as a regulator of VMAT2 function. Removal of the first 16 or 19 AAs of the N-terminus resulted in a molecule with reduced ability to sequester [3H]-5HT. A glutathione-S-transferase-construct of the N-terminus underwent phosphorylation in the presence of PKC at serines 15 and 18. These putative phosphorylation sites were examined for effects on function. Phospho-mimetic substitution of serines 15 and 18 with aspartate in the full-length VMAT2 resulted in reduced [3H]-5HT sequestration and reduced methamphetamine (METH)-stimulated efflux of preloaded [3H]-5HT. In contrast, mutation of serines 15 and 18 to alanines maintained intact net substrate sequestration but eliminated METH-stimulated efflux of pre-accumulated [3H]-5HT. In summary, these data suggest a model in which the VMAT2 N-terminus regulates monoamine sequestration.









![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)