Co-reporter:Stefano Tomassi, Jonas Lategahn, Julian Engel, Marina Keul, Hannah L. Tumbrink, Julia Ketzer, Thomas Mühlenberg, Matthias Baumann, Carsten Schultz-Fademrecht, Sebastian Bauer, and Daniel Rauh
Journal of Medicinal Chemistry March 23, 2017 Volume 60(Issue 6) pp:2361-2361
Publication Date(Web):February 22, 2017
DOI:10.1021/acs.jmedchem.6b01626
The specific targeting of oncogenic mutant epidermal growth factor receptor (EGFR) is a breakthrough in targeted cancer therapy and marks a drastic change in the treatment of non-small cell lung cancer (NSCLC). The recurrent emergence of resistance to these targeted drugs requires the development of novel chemical entities that efficiently inhibit drug-resistant EGFR. Herein, we report the optimization process for a hit compound that has emerged from a phenotypic screen resulting in indazole-based compounds. These inhibitors are conformationally less flexible, target gatekeeper mutated drug-resistant EGFR-L858R/T790M, and covalently alkylate Cys797. Western blot analysis, as well as characterization of the binding kinetics and kinase selectivity profiling, substantiates our approach of targeting drug-resistant EGFR-L858R/T790M with inhibitors incorporating the indazole as hinge binder.
Co-reporter:Steven Smith, Marina Keul, Julian Engel, Debjit Basu, Simone Eppmann, and Daniel Rauh
ACS Omega April 2017? Volume 2(Issue 4) pp:1563-1563
Publication Date(Web):April 20, 2017
DOI:10.1021/acsomega.7b00157
Within the spectrum of kinase inhibitors, covalent-reversible inhibitors (CRIs) provide a valuable alternative approach to classical covalent inhibitors. This special class of inhibitors can be optimized for an extended drug-target residence time. For CRIs, it was shown that the fast addition of thiols to electron-deficient olefins leads to a covalent bond that can break reversibly under proteolytic conditions. Research groups are just beginning to include CRIs in their arsenal of compound classes, and, with that, the understanding of this interesting set of chemical warheads is growing. However, systems to assess both characteristics of the covalent-reversible bond in a simple experimental setting are sparse. Here, we have developed an efficient methodology to characterize the covalent and reversible properties of CRIs and to investigate their potential in targeting clinically relevant variants of the receptor tyrosine kinase EGFR.Topics: Drug discovery and Drug delivery systems; Molecular association; Molecular structure; Proteins; Structure-activity relationship; Structure-activity relationship;
Co-reporter:Mike Bührmann;Julia Hardick;Jörn Weisner;Lena Quambusch; Dr. Daniel Rauh
Angewandte Chemie 2017 Volume 129(Issue 43) pp:13415-13419
Publication Date(Web):2017/10/16
DOI:10.1002/ange.201706345
AbstractEin chemisch-genetischer Ansatz zur kovalenten Adressierung einer einzigartigen lipophilen Bindetasche (LP) in der Mitogen-aktivierten Proteinkinase p38α, deren physiologische Funktion bis heute nicht völlig verstanden ist, wird vorgestellt. Basierend auf einer Reihe von Kokristallstrukturen wurde eine fokussierte Substanzbibliothek von 2-Arylchinazolinen entwickelt, die mit Elektrophilen versehen wurden, um p38α-Mutanten mit artifiziell eingeführten Cysteinen zu generieren. Aufeinander abgestimmte Protein-Liganden-Paare wurden über MS-Analysen identifiziert und mittels MS/MS-basierten sowie kristallographischen Studien validiert. Einige der kovalenten Liganden, die aus diesem Ansatz hervorgingen, zeigten eine hervorragende Selektivität für jeweils eine einzelne p38α-Mutante und können zukünftig als Sondenmoleküle zur Aufklärung der LP-Funktion im Rahmen pharmakologischer Perturbationsexperimente in zellulären Systemen Anwendung finden.
Co-reporter:Julian Engel, Jonas Lategahn, and Daniel Rauh
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 1) pp:2
Publication Date(Web):December 18, 2015
DOI:10.1021/acsmedchemlett.5b00475
In the last five years, the detailed understanding of how to overcome T790M drug resistance in non-small cell lung cancer (NSCLC) has culminated in the development of a third-generation of covalent EGFR inhibitors with excellent clinical outcomes. However, the emergence of a newly discovered acquired drug resistance challenges the concept of small molecule targeted cancer therapy in NSCLC.
Co-reporter:M.Sc. Julian Engel;M.Sc. Christian Becker;M.Sc. Jonas Lategahn;M.Sc. Marina Keul;Julia Ketzer;Dr. Thomas Mühlenberg;M.Sc. Laxmikanth Kollipara;Dr. Carsten Schultz-Fademrecht;Dr. René P. Zahedi;Dr. Sebastian Bauer;Dr. Daniel Rauh
Angewandte Chemie 2016 Volume 128( Issue 36) pp:11069-11073
Publication Date(Web):
DOI:10.1002/ange.201605011
Abstract
Die zielgerichtete Therapie erworbener Wirkstoffresistenz stellt die größte Herausforderung in der Behandlung von EGFR-abhängigem nicht-kleinzelligen Lungenkrebs (NSCLC) dar. Hier berichten wir über die strukturbasierte Entwicklung, Synthese und biologische Evaluierung einer neuen Klasse von kovalenten EGFR-Inhibitoren, die sich durch eine exzellente Inhibition wirkstoffresistenter, EGFR-mutierter Zellen auszeichnet. Röntgenkristallstrukturen in Kombination mit Bindungskinetiken führen zu einem tieferen Verständnis des Inhibitionsmechanismus in EGFR-T790M und liefern Erkenntnisse über Schlüsselaspekte für die effektive Inhibition der kürzlich entdeckten tertiären EGFR-C797S-Mutation.
Co-reporter:Christian Becker;Sinan Öcal;Dr. Hoang D. Nguyen;Dr. Trang Phan;Marina Keul;Dr. Jeffrey R. Simard;Dr. Daniel Rauh
ChemBioChem 2016 Volume 17( Issue 11) pp:990-994
Publication Date(Web):
DOI:10.1002/cbic.201600115
Abstract
The receptor tyrosine kinase EGFR is regulated by complex conformational changes, and this conformational control is disturbed in certain types of cancer. Many ligands are known to bind EGFR in its active conformation, thereby preventing ATP from binding. Only a few ligands are known to stabilize EGFR in its inactive conformation, thus providing novel strategies for perturbing EGFR activity. We report a direct binding assay that enables the identification of novel ligands that bind to and stabilize the inactive conformation of EGFR.
Co-reporter:M.Sc. Julian Engel;M.Sc. Christian Becker;M.Sc. Jonas Lategahn;M.Sc. Marina Keul;Julia Ketzer;Dr. Thomas Mühlenberg;M.Sc. Laxmikanth Kollipara;Dr. Carsten Schultz-Fademrecht;Dr. René P. Zahedi;Dr. Sebastian Bauer;Dr. Daniel Rauh
Angewandte Chemie International Edition 2016 Volume 55( Issue 36) pp:10909-10912
Publication Date(Web):
DOI:10.1002/anie.201605011
Abstract
Targeting acquired drug resistance represents the major challenge in the treatment of EGFR-driven non-small-cell lung cancer (NSCLC). Herein, we describe the structure-based design, synthesis, and biological evaluation of a novel class of covalent EGFR inhibitors that exhibit excellent inhibition of EGFR-mutant drug-resistant cells. Protein X-ray crystallography combined with detailed kinetic studies led to a deeper understanding of the mode of inhibition of EGFR-T790M and provided insight into the key principles for effective inhibition of the recently discovered tertiary mutation at EGFR-C797S.
Co-reporter:Julian Engel; André Richters; Matthäus Getlik; Stefano Tomassi; Marina Keul; Martin Termathe; Jonas Lategahn; Christian Becker; Svenja Mayer-Wrangowski; Christian Grütter; Niklas Uhlenbrock; Jasmin Krüll; Niklas Schaumann; Simone Eppmann; Patrick Kibies; Franziska Hoffgaard; Jochen Heil; Sascha Menninger; Sandra Ortiz-Cuaran; Johannes M. Heuckmann; Verena Tinnefeld; René P. Zahedi; Martin L. Sos; Carsten Schultz-Fademrecht; Roman K. Thomas◆; Stefan M. Kast
Journal of Medicinal Chemistry 2015 Volume 58(Issue 17) pp:6844-6863
Publication Date(Web):August 14, 2015
DOI:10.1021/acs.jmedchem.5b01082
Receptor tyrosine kinases represent one of the prime targets in cancer therapy, as the dysregulation of these elementary transducers of extracellular signals, like the epidermal growth factor receptor (EGFR), contributes to the onset of cancer, such as non-small cell lung cancer (NSCLC). Strong efforts were directed to the development of irreversible inhibitors and led to compound CO-1686, which takes advantage of increased residence time at EGFR by alkylating Cys797 and thereby preventing toxic effects. Here, we present a structure-based approach, rationalized by subsequent computational analysis of conformational ligand ensembles in solution, to design novel and irreversible EGFR inhibitors based on a screening hit that was identified in a phenotype screen of 80 NSCLC cell lines against approximately 1500 compounds. Using protein X-ray crystallography, we deciphered the binding mode in engineered cSrc (T338M/S345C), a validated model system for EGFR-T790M, which constituted the basis for further rational design approaches. Chemical synthesis led to further compound collections that revealed increased biochemical potency and, in part, selectivity toward mutated (L858R and L858R/T790M) vs nonmutated EGFR. Further cell-based and kinetic studies were performed to substantiate our initial findings. Utilizing proteolytic digestion and nano-LC-MS/MS analysis, we confirmed the alkylation of Cys797.
Co-reporter:Daniel Rauh
ACS Chemical Biology 2015 Volume 10(Issue 1) pp:1
Publication Date(Web):January 16, 2015
DOI:10.1021/acschembio.5b00010
Co-reporter:Zhizhou Fang, Jeffrey R. Simard, Dennis Plenker, Hoang D. Nguyen, Trang Phan, Patrik Wolle, Stefan Baumeister, and Daniel Rauh
ACS Chemical Biology 2015 Volume 10(Issue 1) pp:279
Publication Date(Web):June 24, 2014
DOI:10.1021/cb500355c
In addition to the catalytically active kinase domain, most kinases feature regulatory domains that govern their activity. Modulating and interfering with these interdomain interactions presents a major opportunity for understanding biological systems and developing novel therapeutics. Therefore, small molecule inhibitors that target these interactions through an allosteric mode of action have high intrinsic selectivity, as these interactions are often unique to a single kinase or kinase family. Here we report the development of iFLiK (interface-Fluorescent Labels in Kinases), a fluorescence-based assay that can monitor such interdomain interactions. Using iFLiK, we have demonstrated selective detection of allosteric Akt inhibitors that induce an inactive closed conformation unique to Akt. This methodology easily distinguished small molecule allosteric inhibitors from classic ATP-competitive inhibitors. Screening an in-house compound library with iFLiK, we were able to identify novel compounds with a scaffold that has not been previously described for allosteric Akt inhibitors.
Co-reporter:André Richters, Debjit Basu, Julian Engel, Meryem S. Ercanoglu, Hyatt Balke-Want, Roberta Tesch, Roman K. Thomas, and Daniel Rauh
ACS Chemical Biology 2015 Volume 10(Issue 1) pp:289
Publication Date(Web):December 26, 2014
DOI:10.1021/cb500908d
The cytosolic Ser/Thr kinase TBK1 was discovered to be an essential element in the mediation of signals that lead to tumor migration and progression. These findings meet the need for the identification of novel tool compounds and potential therapeutics to gain deeper insights into TBK1 related signaling and its relevance in tumor progression. Herein, we undertake the activity-based screening for unique inhibitors of TBK1 and their subsequent optimization. Initial screening approaches identified a selection of TBK1 inhibitors that were optimized using methods of medicinal chemistry. Variations of the structural characteristics of a representative 2,4,6-substituted pyrimidine scaffold resulted in improved potency. Prospective use as tool compounds or basic contributions to drug design approaches are anticipated for our improved small molecules.
Co-reporter:Svenja C. Mayer-Wrangowski ;Dr. Daniel Rauh
Angewandte Chemie International Edition 2015 Volume 54( Issue 14) pp:4379-4382
Publication Date(Web):
DOI:10.1002/anie.201410148
Abstract
Nuclear receptors are transcription factors that are important targets for current drug discovery efforts as they play a role in many pathological processes. Their activity can be regulated by small molecules like hormones and drugs that can have agonistic or antagonistic functions. These ligands bind to the receptor and account for diverse conformational changes that are crucial determinants for the receptor activity. Here, we set out to develop FLiN (fluorescent labels in nuclear receptors), a direct binding assay that detects conformational changes in the estrogen receptor. The assay is based on the introduction of a cysteine residue and subsequent specific labeling of the receptor with a thiol-reactive fluorophore. Changes in the receptor conformation upon ligand binding lead to differences in the microenvironment of the fluorophore and alter its emission spectrum. The FLiN assay distinguishes between different binding modes and is suitable for high-throughput screening.
Co-reporter:Jörn Weisner;Dr. Rajesh Gontla;Lei vanderWesthuizen;Sebastian Oeck;Julia Ketzer;Dr. Petra Janning;Dr. André Richters;Dr. Thomas Mühlenberg;Dr. Zhizhou Fang;Dr. Abu Taher;Dr. Verena Jendrossek;Dr. Stephen C. Pelly;Dr. Sebastian Bauer;Dr. Willem A. L. vanOtterlo;Dr. Daniel Rauh
Angewandte Chemie International Edition 2015 Volume 54( Issue 35) pp:10313-10316
Publication Date(Web):
DOI:10.1002/anie.201502142
Abstract
Targeting and stabilizing distinct kinase conformations is an instrumental strategy for dissecting conformation-dependent signaling of protein kinases. Herein the structure-based design, synthesis, and evaluation of pleckstrin homology (PH) domain-dependent covalent-allosteric inhibitors (CAIs) of the kinase Akt is reported. These inhibitors bind covalently to a distinct cysteine of the kinase and thereby stabilize the inactive kinase conformation. These modulators exhibit high potency and selectivity, and represent an innovative approach for chemical biology and medicinal chemistry research.
Co-reporter:Jörn Weisner;Dr. Rajesh Gontla;Lei vanderWesthuizen;Sebastian Oeck;Julia Ketzer;Dr. Petra Janning;Dr. André Richters;Dr. Thomas Mühlenberg;Dr. Zhizhou Fang;Dr. Abu Taher;Dr. Verena Jendrossek;Dr. Stephen C. Pelly;Dr. Sebastian Bauer;Dr. Willem A. L. vanOtterlo;Dr. Daniel Rauh
Angewandte Chemie 2015 Volume 127( Issue 35) pp:10452-10456
Publication Date(Web):
DOI:10.1002/ange.201502142
Abstract
Proteinkinasen repräsentieren wichtige Knotenpunkte intrazellulärer Signalwege und sind somit an vielen physiologischen und pathologischen Prozessen beteiligt. Konformations-abhängige Eigenschaften dienen hierbei der Feinregulation ihrer enzymatischen Aktivität und Katalyse-unabhängigen Funktionen. Die Stabilisierung definierter Konformationen ermöglicht detaillierte Analysen solcher Konformations-abhängiger Funktionen. Hier beschreiben wir das strukturbasierte Design, die Synthese und die Charakterisierung Pleckstrin-Homologie(PH)-Domänen-abhängiger Akt-Inhibitoren, die einen neuartigen kovalent-allosterischen Bindungsmodus aufweisen. Durch die kovalente Modifizierung bestimmter Cysteine stabilisieren diese Moleküle die inaktive Kinasekonformation irreversibel. Ihre beträchtliche Inhibitorwirkung und Selektivität bezüglich verwandter Proteinkinasen machen sie zu einem neuartigen Hilfsmittel für die Erforschung chemisch-biologischer und medizinischer Fragestellungen.
Co-reporter:Svenja C. Mayer-Wrangowski ;Dr. Daniel Rauh
Angewandte Chemie 2015 Volume 127( Issue 14) pp:4454-4457
Publication Date(Web):
DOI:10.1002/ange.201410148
Abstract
Kernrezeptoren spielen bei einer Vielzahl physiologischer Prozesse eine entscheidende Rolle und stellen wichtige Zielstrukturen in der modernen Wirkstoff-Forschung dar. Die Aktivität von Kernrezeptoren lässt sich durch niedermolekulare Verbindungen wie Hormone und Wirkstoffe regulieren, die entweder als Agonisten oder als Antagonisten an den Rezeptor binden. Die Bindung dieser Liganden führt zu Konformationsänderungen des Rezeptors, die von entscheidender Bedeutung für dessen Aktivität sind. Das Ziel dieser Arbeit war die Entwicklung einer Methode zur Detektion von Konformationsänderungen des Östrogenrezeptors. Dieses FLiN-Assay-Verfahren (“Fluorescent Labels in Nuclear Receptors”) basiert auf der Einführung eines Cysteins und anschließender Markierung des Rezeptors mit einem Thiol-reaktiven Fluorophor. Konformationsänderungen des Rezeptors führen zu Verschiebungen im Emissionsspektrum des Fluorophors. Neben der Differenzierung von Agonisten und Antagonisten ist FLiN für das Hochdurchsatz-Screening geeignet.
Co-reporter:André Richters ; Hoang D. Nguyen ; Trang Phan ; Jeffrey R. Simard ; Christian Grütter ; Julian Engel
Journal of Medicinal Chemistry 2014 Volume 57(Issue 10) pp:4252-4262
Publication Date(Web):April 22, 2014
DOI:10.1021/jm500167q
Discoidin domain-containing receptors (DDRs) exhibit a unique mechanism of action among the receptor tyrosine kinases (RTKs) because their catalytic activity is induced by extracellular collagen binding. Moreover, they are essential components in the assimilation of extracellular signals. Recently, DDRs were reported to be significantly linked to tumor progression in breast cancer by facilitating the processes of invasion, migration, and metastasis. Here, we report the successful development of a fluorescence-based, direct binding assay for the detection of type II and III DFG-out binders for DDR2. Using sequence alignments and homology modeling, we designed a DDR2 construct appropriate for fluorescent labeling. Successful assay development was validated by sensitive detection of a reference DFG-out binder. Subsequent downscaling led to convenient application to high-throughput screening formats. Screening of a representative compound library identified high-affinity DDR2 ligands validated by orthogonal activity-based assays, and a subset of identified compounds was further investigated with respect to DDR1 inhibition.
Co-reporter:Ralf Schneider ; Claudia Beumer ; Jeffrey R. Simard ; Christian Grütter
Journal of the American Chemical Society 2013 Volume 135(Issue 18) pp:6838-6841
Publication Date(Web):April 23, 2013
DOI:10.1021/ja4030484
Normal cellular function, such as signal transduction, is largely controlled by the reversible phosphorylation of cellular proteins catalyzed by two major classes of enzymes, kinases and phosphatases. A misbalance in this complex and dynamic interplay leads to a variety of severe diseases, such as cancer, inflammation, or autoimmune diseases. This makes kinases as well as phosphatases equally attractive targets for therapeutic manipulation by small molecules. While the development of kinase inhibitors has resulted in several blockbuster drugs, such as imatinib, with remarkable success in the clinic and sales of many billions of U.S. dollars per year, not a single phosphatase inhibitor has yet been approved for clinical use. Similar to the kinase world, substrate-competitive phosphatase inhibitors have been developed but were not suitable for further development into clinical candidates due to their charge and limited selectivity. Research efforts, therefore, have shifted to the exploitation of allosteric sites that can regulate phosphatase activity and may enable the discovery of novel modulators of phosphatase activity with much improved pharmacological properties. However, assay systems, which enable the straightforward discovery of these inhibitor types, are missing. Here, we present a novel binding assay capable of detecting ligands of an allosteric pocket of the protein tyrosine phosphatase 1B. This assay is suitable for high-throughput screening and selectively detects ligands which bind to this unique site with a clear discrimination from substrate-competitive ligands.
Co-reporter:Ralf Schneider ; Anne Gohla ; Jeffrey R. Simard ; Dharmendra B. Yadav ; Zhizhou Fang ; Willem A. L. van Otterlo
Journal of the American Chemical Society 2013 Volume 135(Issue 22) pp:8400-8408
Publication Date(Web):May 14, 2013
DOI:10.1021/ja403074j
In the attempt to discover novel chemical scaffolds that can modulate the activity of disease-associated enzymes, such as kinases, biochemical assays are usually deployed in high-throughput screenings. First-line assays, such as activity-based assays, often rely on fluorescent molecules by measuring a change in the total emission intensity, polarization state, or energy transfer to another fluorescent molecule. However, under certain conditions, intrinsic compound fluorescence can lead to difficult data analysis and to false-positive, as well as false-negative, hits. We have reported previously on a powerful direct binding assay called fluorescent labels in kinases (‘FLiK’), which enables a sensitive measurement of conformational changes in kinases upon ligand binding. In this assay system, changes in the emission spectrum of the fluorophore acrylodan, induced by the binding of a ligand, are translated into a robust assay readout. However, under the excitation conditions of acrylodan, intrinsic compound fluorescence derived from highly conjugated compounds complicates data analysis. We therefore optimized this method by identifying novel fluorophores that excite in the far red, thereby avoiding compound fluorescence. With this advancement, even rigid compounds with multiple π-conjugated ring systems can now be measured reliably. This study was performed on three different kinase constructs with three different labeling sites, each undergoing distinct conformational changes upon ligand binding. It may therefore serve as a guideline for the establishment of novel fluorescence-based detection assays.
Co-reporter:André Richters ; Julia Ketzer ; Matthäus Getlik ; Christian Grütter ; Ralf Schneider ; Johannes M. Heuckmann ; Stefanie Heynck ; Martin L. Sos ; Anu Gupta ▽; Anke Unger ; Carsten Schultz-Fademrecht ; Roman K. Thomas ; Sebastian Bauer
Journal of Medicinal Chemistry 2013 Volume 56(Issue 14) pp:5757-5772
Publication Date(Web):June 17, 2013
DOI:10.1021/jm4004076
Mutations in the catalytic domain at the gatekeeper position represent the most prominent drug-resistant variants of kinases and significantly impair the efficacy of targeted cancer therapies. Understanding the mechanisms of drug resistance at the molecular and atomic levels will aid in the design and development of inhibitors that have the potential to overcome these resistance mutations. Herein, by introducing adaptive elements into the inhibitor core structure, we undertake the structure-based development of type II hybrid inhibitors to overcome gatekeeper drug-resistant mutations in cSrc-T338M, as well as clinically relevant tyrosine kinase KIT-T670I and Abl-T315I variants, as essential targets in gastrointestinal stromal tumors (GISTs) and chronic myelogenous leukemia (CML). Using protein X-ray crystallography, we confirm the anticipated binding mode in cSrc, which proved to be essential for overcoming the respective resistances. More importantly, the novel compounds effectively inhibit clinically relevant gatekeeper mutants of KIT and Abl in biochemical and cellular studies.
Co-reporter:Zhizhou Fang, Christian Grütter, and Daniel Rauh
ACS Chemical Biology 2013 Volume 8(Issue 1) pp:58
Publication Date(Web):December 18, 2012
DOI:10.1021/cb300663j
The modulation of kinase function has become an important goal in modern drug discovery and chemical biology research. In cancer-targeted therapies, kinase inhibitors have been experiencing an upsurge, which can be measured by the increasing number of kinase inhibitors approved by the FDA in recent years. However, lack of efficacy, limited selectivity, and the emergence of acquired drug resistance still represent major bottlenecks in the clinic and challenge inhibitor development. Most known kinase inhibitors target the active kinase and are ATP competitive. A second class of small organic molecules, which address remote sites of the kinase and stabilize enzymatically inactive conformations, is rapidly moving to the forefront of kinase inhibitor research. Such allosteric modulators bind to sites that are less conserved across the kinome and only accessible upon conformational changes. These molecules are therefore thought to provide various advantages such as higher selectivity and extended drug target residence times. This review highlights various strategies that have been developed to utilizing exclusive structural features of kinases and thereby modulating their activity allosterically.
Co-reporter:Ralf Schneider ; Christian Becker ; Jeffrey R. Simard ; Matthäus Getlik ; Nina Bohlke ; Petra Janning
Journal of the American Chemical Society 2012 Volume 134(Issue 22) pp:9138-9141
Publication Date(Web):May 21, 2012
DOI:10.1021/ja303858w
Abelson (Abl) tyrosine kinase is an important cellular enzyme that is rendered constitutively active in the breakpoint cluster region (BCR)-Abl fusion protein, contributing to several forms of leukemia. Although inhibiting BCR-Abl activity with imatinib shows great clinical success, many patients acquire secondary mutations that result in resistance to imatinib. Second-generation inhibitors such as dasatinib and nilotinib can overcome the majority of these mutations but fail to treat patients with an especially prevalent T315I mutation at the gatekeeper position of the kinase domain. However, a combination of nilotinib with an allosteric type IV inhibitor was recently shown to overcome this clinically relevant point mutation. In this study, we present the development of a direct binding assay that enables the straightforward detection of allosteric inhibitors which bind within the myristate pocket of Abl. The assay is amenable to high-throughput screening and exclusively detects the binding of ligands to this unique allosteric site.
Co-reporter:André Koch, Haridas B. Rode, André Richters, Daniel Rauh, and Silke Hauf
ACS Chemical Biology 2012 Volume 7(Issue 4) pp:723
Publication Date(Web):January 20, 2012
DOI:10.1021/cb200465c
The perturbation of protein kinases with small organic molecules is a powerful approach to dissect kinase function in complex biological systems. Covalent kinase inhibitors that target thiols in the ATP binding pocket of the kinase domain proved to be ideal reagents for the investigation of highly dynamic cellular processes. However, due to the covalent inhibitors' possible off-target reactivities, it is required that the overall shape of the inhibitor as well as the intrinsic reactivity of the electrophile are precisely tuned to favor the reaction with only the desired cysteine. Here we report on the design and biological characterization of covalent anilinoquinazolines as potent inhibitors of genetically engineered Aurora kinase in fission yeast.
Co-reporter:Christian Grütter, Jeffrey R. Simard, Svenja C. Mayer-Wrangowski, Peter H. Schreier, José Pérez-Martín, André Richters, Matthäus Getlik, Oliver Gutbrod, Christoph A. Braun, Michael E. Beck, and Daniel Rauh
ACS Chemical Biology 2012 Volume 7(Issue 7) pp:1257
Publication Date(Web):April 30, 2012
DOI:10.1021/cb300128b
Protein kinases are key enzymes in the complex regulation of cellular processes in almost all living organisms. For this reason, protein kinases represent attractive targets to stop the growth of eukaryotic pathogens such as protozoa and fungi. However, using kinase inhibitors to fight against these organisms bears several challenges since most of them are unselective and will also affect crucial host kinases. Here we present the X-ray structure of glycogen synthase kinase 3 from the fungal plant pathogen Ustilago maydis (UmGSK3) and its inhibition by type-II kinase inhibitors. Despite the high sequence homology between the human and the fungal variant of this vital kinase, we found substantial differences in the conformational plasticity of their active sites. Compounds that induced such conformational changes could be used to selectively inhibit the fungal kinase. This study serves as an example of how species-specific selectivity of inhibitors can be achieved by identifying and addressing the inactive state of a protein kinase. In addition to this, our study gives interesting insights into the molecular plasticity of UmGSK3 by revealing a previously unknown inactive conformation of this important kinase family.
Co-reporter:Matthäus Getlik, Christian Grütter, Jeffrey R. Simard, Hoang D. Nguyen, Armin Robubi, Beate Aust, Willem A.L. van Otterlo, Daniel Rauh
European Journal of Medicinal Chemistry 2012 Volume 48() pp:1-15
Publication Date(Web):February 2012
DOI:10.1016/j.ejmech.2011.11.019
In this paper, we present the structure-based design, synthesis and biological activity of N-pyrazole, N′-thiazole-ureas as potent inhibitors of p38α mitogen-activated protein kinase (p38α MAPK). Guided by complex crystal structures, we employed the initially identified N-aryl, N′-thiazole urea scaffold and introduced key structural elements that allowed the formation of novel hydrogen bonding interactions within the allosteric site of p38α, resulting in potent type III inhibitors. [4-(3-tert-Butyl-5-{[(1,3-thiazol-2-ylamino)carbonyl]amino}-1H-pyrazol-1-yl)-phenyl]acetic acid 18c was found to be the most potent compound within this series and inhibited p38α activity with an IC50 of 135 ± 21 nM. Its closest analog, ethyl [4-(3-tert-butyl-5-{[(1,3-thiazol-2-ylamino)carbonyl]amino}-1H-pyrazol-1-yl)phenyl]acetate 18b, effectively inhibited p38α mediated phosphorylation of the mitogen activated protein kinase activated protein kinase 2 (MK2) in HeLa cells.The SAR of a series of N-pyrazole, N′-thiazole-ureas was explored systematically and resulted in ethyl [4-(3-tert-butyl-5-{[(1,3-thiazol-2-ylamino)carbonyl]amino}-1H-pyrazol-1-yl)phenyl]acetate (18b) with potent cellular activity against p38α. Highlights► FLiK assay technology to identify stabilizers of enzymatically inactive p38α. ► Protein X-ray crystallography of screening hits. ► Novel binding mode of screening hit. ► Structure-based design approaches to improve inhibitor potency.
Co-reporter:Ruth Brenk, Daniel Rauh
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 12) pp:3695-3697
Publication Date(Web):15 June 2012
DOI:10.1016/j.bmc.2012.04.038
Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Svenja Mayer-Wrangowski
BIOspektrum 2012 Volume 18( Issue 4) pp:376-379
Publication Date(Web):2012 June
DOI:10.1007/s12268-012-0195-7
Selective kinase inhibitors have become an important class of anti-cancer agents. However, clinical success of these agents is mostly limited to a small subset of patients, who are often defined by specific genomic lesions within their tumor cells. Recent progress in cancer genetics and medicinal chemistry allows for the first time effective personalization of tumor therapy with so far unreached benefit for some patient populations.
Co-reporter:Sandra Tückmantel, Jörg N. Greul, Petra Janning, Andreas Brockmeyer, Christian Grütter, Jeffrey R. Simard, Oliver Gutbrod, Michael E. Beck, Klaus Tietjen, Daniel Rauh, and Peter H. Schreier
ACS Chemical Biology 2011 Volume 6(Issue 9) pp:926
Publication Date(Web):June 14, 2011
DOI:10.1021/cb200112y
Infestation of crops by pathogenic fungi has continued to have a major impact by reducing yield and quality, emphasizing the need to identify new targets and develop new agents to improve methods of crop protection. Here we present Aurora kinase from the phytopathogenic fungus Ustilago maydis as a novel target for N-substituted diaminopyrimidines, a class of small-molecule kinase inhibitors. We show that Aurora kinase is essential in U. maydis and that diaminopyrimidines inhibit its activity in vitro. Furthermore, we observed an overall good correlation between in vitro inhibition of Aurora kinase and growth inhibition of diverse fungi in vivo. In vitro inhibition assays with Ustilago and human Aurora kinases indicate that some compounds of the N-substituted diaminopyrimidine class show specificity for the Ustilago enzyme, thus revealing their potential as selective fungicides.
Co-reporter:Viktor V. Vintonyak, Herbert Waldmann, Daniel Rauh
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 7) pp:2145-2155
Publication Date(Web):1 April 2011
DOI:10.1016/j.bmc.2011.02.047
The site specific functionalization of phosphate groups with amino acid side chains of substrate proteins represents one of the most important regulatory mechanisms of biological systems. Phosphorylation and dephosphorylation are reversibly catalyzed by protein kinases and protein phosphatases, and the aberrant regulation of these enzymes is associated with the onset and progression of various disease states such as cancer, diabetes, neurodegenerative and autoimmune disorders, making these proteins attractive targets for drug discovery. Here we report on strategies currently explored for the identification and development of various inhibitors directed against clinically relevant phosphatases. While over the last years, inhibition of phosphorylation has evolved into a key strategy in targeted therapies, the development of clinically relevant phosphatase inhibitors still faces major bottlenecks and is often plagued by limited selectivity and unfavorable pharmacokinetics. The reader will gain a better understanding of the importance of the field and its current limitations.
Co-reporter:Christian Grütter, Shivakumar Sreeramulu, Guido Sessa, Daniel Rauh
Journal of Molecular Biology (15 November 2013) Volume 425(Issue 22) pp:4455-4467
Publication Date(Web):15 November 2013
DOI:10.1016/j.jmb.2013.07.034
•First insights into the structural architecture of a BSK family member.•Identification of unusual conformational arrangements within the active site.•Structural and sequence comparisons imply that BSKs are pseudokinases.•Activity test reveal that BSKs are catalytically inactive protein kinases.Brassinosteroid signaling kinases (BSKs) are plant-specific receptor-like cytoplasmic protein kinases involved in the brassinosteroid signaling pathway. Unlike common protein kinases, they possess a naturally occurring alanine residue at the “gatekeeper” position, as well as other sequence variations. How BSKs activate downstream proteins such as BSU1, as well as the structural consequences of their unusual sequential features, was unclear. We crystallized the catalytic domain of BSK8 and solved its structure by multiple-wavelength anomalous dispersion phasing methods to a resolution of 1.5 Å. In addition, a co-crystal structure of BSK8 with 5-adenylyl imidodiphosphate (AMP-PNP) revealed unusual conformational arrangements of the nucleotide phosphate groups and catalytic key motifs, typically not observed for active protein kinases. Sequential analysis and comparisons with known pseudokinase structures suggest that BSKs represent constitutively inactive protein kinases that regulate brassinosteroid signal transfer through an allosteric mechanism.Download high-res image (298KB)Download full-size image

![1-Piperidinecarboxylic acid, 3-(4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-, 1,1-dimethylethyl ester, (3R)-](/data/chemimg/2268000/1276110-38-3.png)
![1-Piperidinecarboxylic acid, 3-(4-amino-3-iodo-1H-pyrazolo[3,4-d]pyrimidin-1-yl)-, 1,1-dimethylethyl ester, (3R)-](/data/chemimg/2268000/1276110-38-3_b.png)
![Benzamide,3-fluoro-5-(4-morpholinyl)-N-[4-[2-(4-morpholinyl)ethoxy]-1-naphthalenyl]-](http://img.cochemist.com/ccimg/874200/874132-77-1.png)
![Benzamide,3-fluoro-5-(4-morpholinyl)-N-[4-[2-(4-morpholinyl)ethoxy]-1-naphthalenyl]-](http://img.cochemist.com/ccimg/874200/874132-77-1_b.png)



![1,6-Naphthyridine, 5-chloro-2-[4-(1,3-dioxolan-2-yl)phenyl]-3-phenyl-](http://img.cochemist.com/ccimg/917400/917363-82-7.png)
![1,6-Naphthyridine, 5-chloro-2-[4-(1,3-dioxolan-2-yl)phenyl]-3-phenyl-](http://img.cochemist.com/ccimg/917400/917363-82-7_b.png)
![4-(4-chlorobenzyl)-1-(7H-pyrrolo[2,3-d]pyriMidin-4-yl)piperidin-4-aMine](http://img.cochemist.com/ccimg/885500/885499-61-6.png)
![4-(4-chlorobenzyl)-1-(7H-pyrrolo[2,3-d]pyriMidin-4-yl)piperidin-4-aMine](http://img.cochemist.com/ccimg/885500/885499-61-6_b.png)
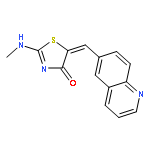
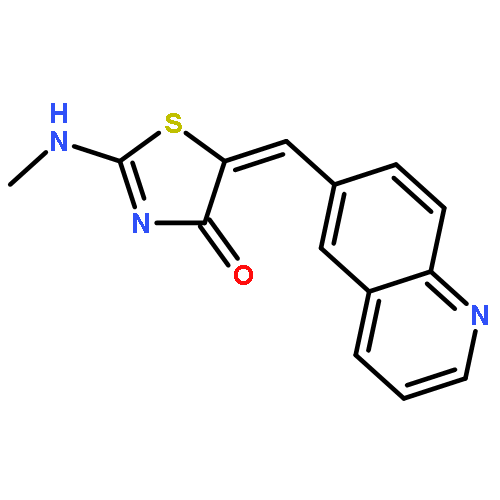
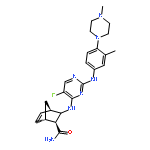
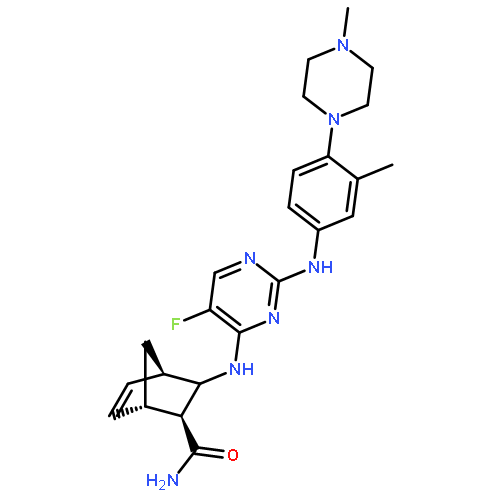
![Ethanone, 1-[4-(1,3-dioxolan-2-yl)phenyl]-2-phenyl-](http://img.cochemist.com/ccimg/868300/868280-61-9.png)
![Ethanone, 1-[4-(1,3-dioxolan-2-yl)phenyl]-2-phenyl-](http://img.cochemist.com/ccimg/868300/868280-61-9_b.png)
![7-(4-Methylphenyl)-5-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-ol](http://img.cochemist.com/ccimg/865600/865546-60-7.png)
![7-(4-Methylphenyl)-5-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-ol](http://img.cochemist.com/ccimg/865600/865546-60-7_b.png)
![7-(3-Methylphenyl)-5-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-ol](http://img.cochemist.com/ccimg/865600/865546-58-3.png)
![7-(3-Methylphenyl)-5-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-ol](http://img.cochemist.com/ccimg/865600/865546-58-3_b.png)











![2-METHYL-5-NITRO-1-BENZENESULFONIC ACID 2-[(6-BROMOIMIDAZO[1,2-A]PYRIDIN-3-YL)METHYLENE]-1-METHYLHYDRAZIDE](http://img.cochemist.com/ccimg/372200/372196-67-3.png)
![2-METHYL-5-NITRO-1-BENZENESULFONIC ACID 2-[(6-BROMOIMIDAZO[1,2-A]PYRIDIN-3-YL)METHYLENE]-1-METHYLHYDRAZIDE](http://img.cochemist.com/ccimg/372200/372196-67-3_b.png)









![1H-Pyrrole-2,5-dione,3-[(3,5-dichloro-4-hydroxyphenyl)amino]-4-(3-nitrophenyl)-](http://img.cochemist.com/ccimg/264300/264211-21-4.png)
![1H-Pyrrole-2,5-dione,3-[(3,5-dichloro-4-hydroxyphenyl)amino]-4-(3-nitrophenyl)-](http://img.cochemist.com/ccimg/264300/264211-21-4_b.png)
![Urea,N-[3-(1,1-dimethylethyl)-1-(4-methylphenyl)-1H-pyrazol-5-yl]-N'-1-naphthalenyl-](http://img.cochemist.com/ccimg/223800/223724-68-3.png)
![Urea,N-[3-(1,1-dimethylethyl)-1-(4-methylphenyl)-1H-pyrazol-5-yl]-N'-1-naphthalenyl-](http://img.cochemist.com/ccimg/223800/223724-68-3_b.png)
![7-(4-Methoxyphenyl)-5-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-ol](http://img.cochemist.com/ccimg/220900/220835-17-6.png)
![7-(4-Methoxyphenyl)-5-phenyl-7H-pyrrolo[2,3-d]pyrimidin-4-ol](http://img.cochemist.com/ccimg/220900/220835-17-6_b.png)




![Carbamimidothioic acid,3-[3-[2,5-dihydro-4-(1-methyl-1H-indol-3-yl)-2,5-dioxo-1H-pyrrol-3-yl]-1H-indol-1-yl]propylester](http://img.cochemist.com/ccimg/125400/125314-64-9.png)
![Carbamimidothioic acid,3-[3-[2,5-dihydro-4-(1-methyl-1H-indol-3-yl)-2,5-dioxo-1H-pyrrol-3-yl]-1H-indol-1-yl]propylester](http://img.cochemist.com/ccimg/125400/125314-64-9_b.png)
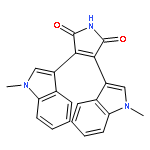
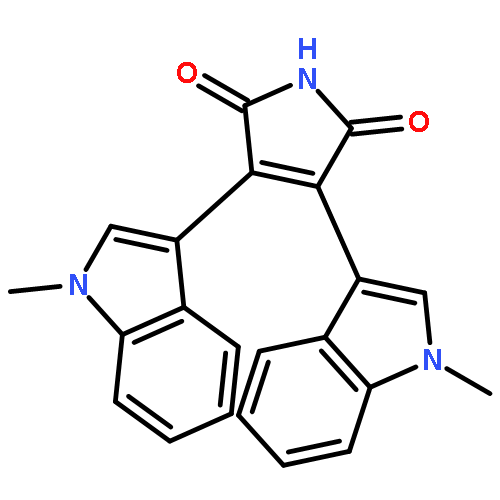
![1H-Pyrrole-2,5-dione,3-[1-(3-aminopropyl)-1H-indol-3-yl]-4-(1-methyl-1H-indol-3-yl)-](http://img.cochemist.com/ccimg/125400/125313-65-7.png)
![1H-Pyrrole-2,5-dione,3-[1-(3-aminopropyl)-1H-indol-3-yl]-4-(1-methyl-1H-indol-3-yl)-](http://img.cochemist.com/ccimg/125400/125313-65-7_b.png)
![3-[1-(3-Hydroxypropyl)-1H-indol-3-yl]-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione](http://img.cochemist.com/ccimg/125400/125313-60-2.png)
![3-[1-(3-Hydroxypropyl)-1H-indol-3-yl]-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione](http://img.cochemist.com/ccimg/125400/125313-60-2_b.png)
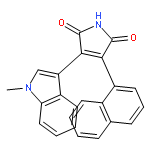
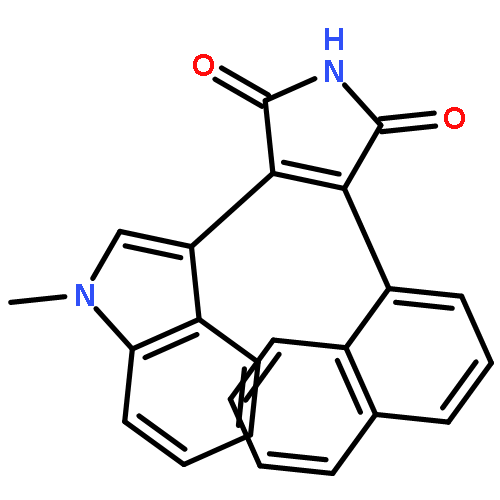
![5H-Indolo[2,3-a]pyrrolo[3,4-c]carbazole-5,7(6H)-dione,12,13-dihydro-3,9-dimethoxy-](http://img.cochemist.com/ccimg/118500/118458-58-5.png)
![5H-Indolo[2,3-a]pyrrolo[3,4-c]carbazole-5,7(6H)-dione,12,13-dihydro-3,9-dimethoxy-](http://img.cochemist.com/ccimg/118500/118458-58-5_b.png)
![9,12-Epoxy-1H-diindolo[1,2,3-fg:3',2',1'-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylicacid, 2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-, methyl ester,(9S,10R,12R)-](http://img.cochemist.com/ccimg/99600/99533-80-9.png)
![9,12-Epoxy-1H-diindolo[1,2,3-fg:3',2',1'-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylicacid, 2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-, methyl ester,(9S,10R,12R)-](http://img.cochemist.com/ccimg/99600/99533-80-9_b.png)


![2-[methyl(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]ethyl iodoacetate](/data/chemimg/1486800/67013-48-3.png)
![2-[methyl(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]ethyl iodoacetate](/data/chemimg/1486800/67013-48-3_b.png)


![Benzonitrile,4-imidazo[1,2-a]pyridin-2-yl-](http://img.cochemist.com/ccimg/55900/55843-91-9.png)
![Benzonitrile,4-imidazo[1,2-a]pyridin-2-yl-](http://img.cochemist.com/ccimg/55900/55843-91-9_b.png)
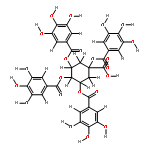
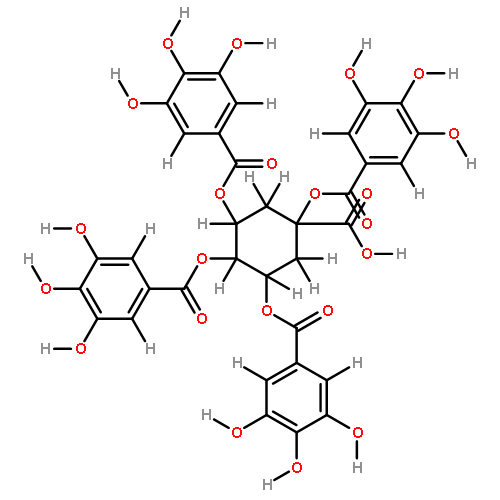
![Benzoic acid,3,4,5-trihydroxy-,(2R,3R)-2-[1-[(2R,3R)-3,4-dihydro-3,5,7-trihydroxy-2H-1-benzopyran-2-yl]-3,4,6-trihydroxy-5-oxo-5H-benzocyclohepten-8-yl]-3,4-dihydro-5,7-dihydroxy-2H-1-benzopyran-3-ylester](http://img.cochemist.com/ccimg/30500/30462-34-1.png)
![Benzoic acid,3,4,5-trihydroxy-,(2R,3R)-2-[1-[(2R,3R)-3,4-dihydro-3,5,7-trihydroxy-2H-1-benzopyran-2-yl]-3,4,6-trihydroxy-5-oxo-5H-benzocyclohepten-8-yl]-3,4-dihydro-5,7-dihydroxy-2H-1-benzopyran-3-ylester](http://img.cochemist.com/ccimg/30500/30462-34-1_b.png)




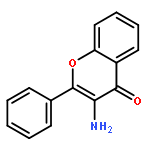
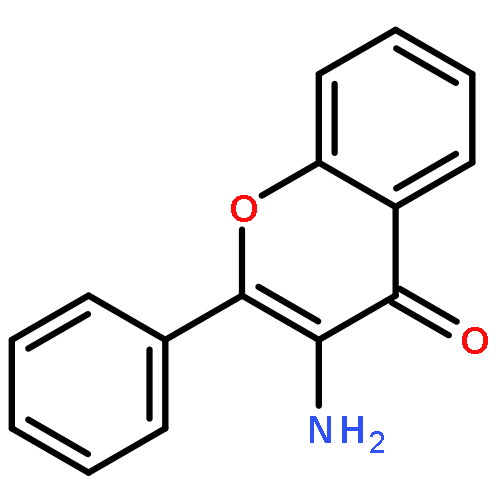

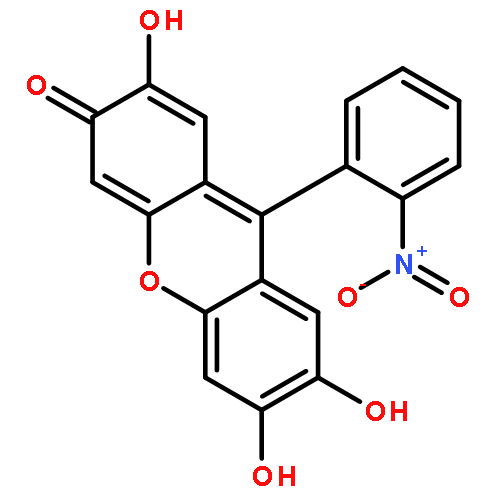
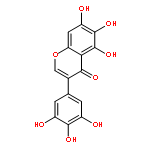







![N-[3-[5-CHLORO-2-[2-METHOXY-4-(4-METHYLPIPERAZIN-1-YL)ANILINO]PYRIMIDIN-4-YL]OXYPHENYL]PROP-2-ENAMIDE](http://img.cochemist.com/ccimg/1213300/1213269-23-8.png)
![N-[3-[5-CHLORO-2-[2-METHOXY-4-(4-METHYLPIPERAZIN-1-YL)ANILINO]PYRIMIDIN-4-YL]OXYPHENYL]PROP-2-ENAMIDE](http://img.cochemist.com/ccimg/1213300/1213269-23-8_b.png)


![(S)-4-(2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-(piperidin-3-ylmethoxy)-1H-imidazo[4,5-c]pyridin-4-yl)-2-methylbut-3-yn-2-ol](http://img.cochemist.com/ccimg/937200/937174-76-0.png)
![(S)-4-(2-(4-Amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-(piperidin-3-ylmethoxy)-1H-imidazo[4,5-c]pyridin-4-yl)-2-methylbut-3-yn-2-ol](http://img.cochemist.com/ccimg/937200/937174-76-0_b.png)



![4-amino-5-fluoro-3-[5-(4-methylpiperazin-1-yl)-1h-benzimidazol-2-yl]quinolin-2(1h)-one](http://img.cochemist.com/ccimg/405200/405169-16-6.png)
![4-amino-5-fluoro-3-[5-(4-methylpiperazin-1-yl)-1h-benzimidazol-2-yl]quinolin-2(1h)-one](http://img.cochemist.com/ccimg/405200/405169-16-6_b.png)