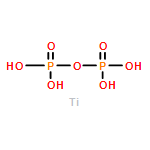
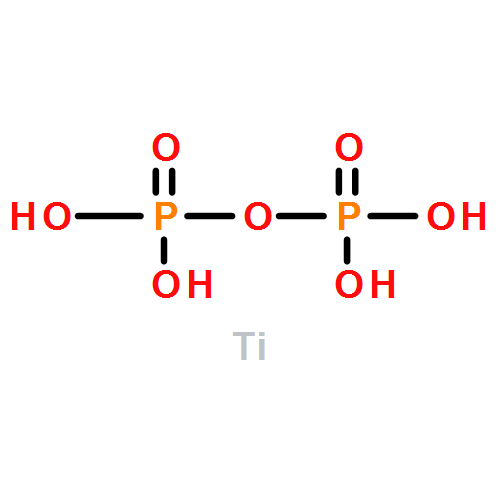
Co-reporter:Tingting Liu, Xiao Ma, Danni Liu, Shuai Hao, Gu Du, Yongjun Ma, Abdullah M. Asiri, Xuping Sun, and Liang Chen
ACS Catalysis January 6, 2017 Volume 7(Issue 1) pp:98-98
Publication Date(Web):November 23, 2016
DOI:10.1021/acscatal.6b02849
Heteratom doping is a possible way to tune the hydrogen evolution reaction (HER) catalytic capability of electrocatalysts. In this work, we report the development of Mn-doped CoP (Mn–Co–P) nanosheets array on Ti mesh (Mn–Co–P/Ti) as an efficient 3D HER electrocatalyst with good stability at all pH values. Electrochemical tests demonstrate that Mn doping leads to enhanced catalytic activity of CoP. In 0.5 M H2SO4, this Mn–Co–P/Ti catalyst drives 10 mA cm–2 at an overpotential of 49 mV, which is 32 mV less than that for CoP/Ti. To achieve the same current density, it demands overpotentials of 76 and 86 mV in 1.0 M KOH and phosphate-buffered saline, respectively. The enhanced HER activity for Mn–Co–P can be attributed to its more thermo-neutral hydrogen adsorption free energy than CoP, which is supported by density functional theory calculations.Keywords: all pH values; CoP; electrocatalysts; hydrogen evolution reaction; Mn doping;
Co-reporter:Li Yang;Danni Liu;Shuai Hao;Fengli Qu;Ruixiang Ge;Yongjun Ma;Gu Du;Abdullah M. Asiri;Xuping Sun
Analytical Chemistry February 21, 2017 Volume 89(Issue 4) pp:2191-2195
Publication Date(Web):January 31, 2017
DOI:10.1021/acs.analchem.6b04760
Nanostructures possess distinct quenching ability toward fluorophores with different emission frequencies and have been intensively used as nanoquenchers for homogeneous nucleic acid detection. Complete understanding of such a sensing system will provide significant guidance for the design of superior sensing materials, which is still lacking. In this Letter, we demonstrate the development of FeP nanowires as a nanoquencher for high-performance fluorescent nucleic acid detection with much superior performance to α-Fe2O3 counterparts. The whole detection process is complete within 1 min, and this fluorosensor presents a detection limit as low as 4 pM with strong discrimination of single-point mutation. Electrochemical tests and density functional theory calculations reveal that FeP NWs are superior in both conductivity for facilitated electron diffusion and hydrogen-evolving catalytic activity for favorable electron depletion, providing further experimental and theoretical insights into the enhanced sensing performance of the FeP nanosensor. Both faster electron transfer kinetics and stronger electron-consuming ability via catalyzed proton reduction enable FeP nanowires to be a superb nucleic acid nanosensor for applications.
Co-reporter:Mitchell R. Armstrong, Sethuraman Senthilnathan, Christopher J. Balzer, Bohan Shan, Liang Chen, Bin Mu
Ultrasonics Sonochemistry 2017 Volume 34() pp:365-370
Publication Date(Web):January 2017
DOI:10.1016/j.ultsonch.2016.06.011
•A competitive mechanism for MOF growth in sonochemical reactor is proposed.•The mechanism includes constructive crystal growth and deconstructive sonofragmentation.•Sonication amplitude and solvent choice are primary factors determining the particle size of HKUST-1.•Sonicator tip size and reactor size are primary factors contributing to particle size distribution.•Sonication time has a significant effect on yield.Systematic studies of key operating parameters for the sonochemical synthesis of the metal organic framework (MOF) HKUST-1(also called CuBTC) were performed including reaction time, reactor volume, sonication amplitude, sonication tip size, solvent composition, and reactant concentrations analyzed through SEM particle size analysis. Trends in the particle size and size distributions show reproducible control of average particle sizes between 1 and 4 μm. These results along with complementary studies in sonofragmentation and temperature control were conducted to compare these results to kinetic crystal growth models found in literature to develop a plausible hypothetical mechanism for ultrasound-assisted growth of metal-organic-frameworks composed of a competitive mechanism including constructive solid-on-solid (SOS) crystal growth and a deconstructive sonofragmentation.
Co-reporter:Bai-Hai LiYun Lin, Jian-Lin Wang, Xue Zhang, Yun-Rui Wang, Yu Jiang, Ting-Shuai Li, Li-Min Liu, Liang ChenWan-Li Zhang, Yan-Rong Li
The Journal of Physical Chemistry C 2017 Volume 121(Issue 1) pp:
Publication Date(Web):December 7, 2016
DOI:10.1021/acs.jpcc.6b07509
Designing highly efficient photocatalysts using first-principles calculations is an urgent challenge. In this work, the potential structures of doped bronze TiO2 were systematically designed and screened using cluster expansion (CE) and first-principles calculations. The ordered TiPtO4 phase, which can be fabricated by substituting the Ti cations in bronze TiO2 with Pt, is predicted to be a promising visible light responsive photocatalyst with excellent thermodynamic stability. Further calculations suggest that TiPtO4 is a semiconductor with a suitable band gap (1.96 eV) for the absorption of visible light and appropriate band edge positions relative to the redox potential of water splitting. This work not only reports a potential highly efficient material for photocatalysis but also sheds light on the design and rationalization of this new photocatalyst using first-principles calculations.
Co-reporter:Ling Zhang;Lisi Xie;Min Ma;Fengli Qu;Gu Du;Abdullah M. Asiri;Xuping Sun
Catalysis Science & Technology (2011-Present) 2017 vol. 7(Issue 13) pp:2689-2694
Publication Date(Web):2017/07/03
DOI:10.1039/C7CY00703E
Designing and developing non-noble-metal electrocatalysts for efficient full water splitting at neutral pH is highly desired but still remains a huge challenge. In this communication, we report the development of homologous Co-based nanowire films, Co2N nanowire film on a Ti mesh (Co2N/TM) and Co-phosphate nanowire film on a Ti mesh (Co–Pi/TM), as complementary catalysts for stable electrochemical hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) in neutral electrolyte. Co2N/TM shows remarkable HER activity with the need of an overpotential of 290 mV to drive 10 mA cm−2 in 1.0 M phosphate-buffered saline (PBS), and Co–Pi/TM is superior in OER activity affording 10 mA cm−2 at overpotentials of only 430 and 300 mV in 0.1 and 1.0 M PBS, respectively. The two-electrode water electrolyzer using Co2N/TM as a cathode and Co–Pi/TM as an anode affords a water-splitting current density of 10 mA cm−2 at a cell voltage of 1.78 V with 100% Faradaic efficiency in 1.0 M PBS, promising the practical uses of such homologous Co-based catalyst materials in technological devices.
Co-reporter:Tingting Liu;Danni Liu;Fengli Qu;Dengxing Wang;Ling Zhang;Ruixiang Ge;Shuai Hao;Yongjun Ma;Gu Du;Abdullah M. Asiri;Xuping Sun
Advanced Energy Materials 2017 Volume 7(Issue 15) pp:
Publication Date(Web):2017/08/01
DOI:10.1002/aenm.201700020
As a non-toxic species, Zn fulfills a multitude of biological roles, but its promoting effect on electrocatalysis has been rarely explored. Herein, the theoretic predications and experimental investigations that nonelectroactive Zn behaves as an effective promoter for CoP-catalyzed hydrogen evolution reaction (HER) in both acidic and alkaline media is reported. Density function theory calculations reveal that Zn doing leads to more thermal-neutral hydrogen adsorption free energy and thus enhanced HER activity for CoP catalyst. Electrochemical tests show that a Zn0.08Co0.92P nanowall array on titanium mesh (Zn0.08Co0.92P/TM) needs overpotentials of only 39 and 67 mV to drive a geometrical catalytic current of 10 mA cm-2 in 0.5 m H2SO4 and 1.0 m KOH, respectively. This Zn0.08Co0.92P/TM is also superior in activity over CoP/TM for urea oxidation reaction (UOR), driving 115 mA cm-2 at 0.6 V in 1.0 m KOH with 0.5 m urea. The high HER and UOR activity of this bifunctional electrode enables a Zn0.08Co0.92P/TM-based two-electrode electrolyzer for energy-saving hydrogen production, offering 10 mA cm-2 at a low voltage of 1.38 V with strong long-term electrochemical stability.
Co-reporter:Jianhui Yang, Xuepiao Luo, Xumeng Zhou, Shaozheng Zhang, Jia Liu, Yan Xie, Liang Lv, Liang Chen
Computational Materials Science 2017 Volume 139(Volume 139) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.commatsci.2017.08.016
•Density functional theory calculations show that Cr2M2C3T2 systems are magnetic.•Cr2Ti2C3O2 and Cr2V2C3O2 are ferromagnetic in strain-free state.•Ferromagnetic to anti-ferromagnetic (or anti-ferromagnetic to ferromagnetic) phase transitions occurred for Cr2Ti2C3T2 and Cr2V2C3T2 systems under −5% to 5% extensile strains, but not Cr2Ti2C3O2.•The magnetic moment of Cr atoms increases monotonically as extensile strain increases from −5% to 5% for all Cr2M2C3T2 systems.The influence of extensile strain on the magnetic properties of Cr2M2C3T2 (M = Ti, V, Nb, and Ta; T = OH, O, and F) are investigated using density functional theory. Calculation results show that the ferromagnetic arrangement is energetically favorable for Cr2Ti2C3O2 and Cr2V2C3O2 in the strain-free state. The Curie temperatures are 720.6 K and 246.8 K, respectively, calculated using the Heisenberg model with mean-field approximation. Other systems are anti-ferromagnetic with magnetic moment of Cr atoms (MCr) larger than 1.9 μB. For extensile strains that vary from −5% to 5%, the anti-ferromagnetic arrangement is always energetically favorable for Cr2Nb2C3T2 and Cr2Nb2C3T2 systems. However, ferromagnetic to anti-ferromagnetic (or anti-ferromagnetic to ferromagnetic) phase transitions occurred for Cr2Ti2C3T2 and Cr2V2C3T2, but not Cr2Ti2C3O2. The magnetic moment of Cr atoms increases monotonically as extensile strain increases from −5% to 5% for all Cr2M2C3T2 systems. The projected density of states shows that Cr-3d orbitals become more localized as the strain increases, and that the density of states near the fermi level decreases, which may reduce the conductivity. This study indicates that the magnetic and electronic properties of Cr2M2C3T2 (where M = Ti, or V; T = O, OH, or F) can be effectively tuned using extensile strain. This promotes the application of these two-dimensional materials in spin electronics and data storage.Download high-res image (180KB)Download full-size image
Co-reporter:Chunlong Kong;Hongbing Du;Banglin Chen
Energy & Environmental Science (2008-Present) 2017 vol. 10(Issue 8) pp:1812-1819
Publication Date(Web):2017/08/09
DOI:10.1039/C7EE00830A
In this study, we present a general and efficient strategy to design and prepare novel MOF-based hybrid membranes on a tubular ceramic substrate. Thin and compact MOF/organosilica nano-composite membranes can be rationally formed and tightly bound with the tubular ceramic substrate. The resulting membranes display high gas separation performance with both high selectivity and high permeation rate, which can be well correlated to the parent MOFs. The as-prepared ZIF-8 and MIL-53-NH2 incorporated organosilica nanocomposite membranes have been demonstrated as some of the best performing MOF-based membranes for highly H2- (H2/CH4 selectivity = 26.5, H2 permeance = 1.06 × 10−6 mol m−2 s−1 Pa−1) and CO2-selective (CO2/CH4 selectively = 18.2, CO2 permeance = 1.44 × 10−7 mol m−2 s−1 Pa−1) separations, for H2/CH4 (1 : 1) and CO2/CH4 (1 : 1) mixtures at room temperature, respectively.
Co-reporter:Lisi Xie;Fengli Qu;Zhiang Liu;Xiang Ren;Shuai Hao;Ruixiang Ge;Gu Du;Abdullah M. Asiri;Xuping Sun
Journal of Materials Chemistry A 2017 vol. 5(Issue 17) pp:7806-7810
Publication Date(Web):2017/05/03
DOI:10.1039/C7TA02333B
It is of great importance but still remains a key challenge to develop non-noble-metal bifunctional catalysts for efficient full water splitting under mild pH conditions. In this communication, we report the in situ electrochemical development of an ultrathin Ni–Bi layer on a metallic Ni3N nanosheet array supported on a Ti mesh (Ni3N@Ni–Bi NS/Ti) as a durable 3D core/shell structured nanoarray electrocatalyst for water oxidation at near-neutral pH. The Ni3N@Ni–Bi NS/Ti demands overpotentials of 405 and 382 mV to deliver a geometrical catalytic current density of 10 mA cm−2 in 0.1 and 0.5 M K–Bi (pH: 9.2), respectively, superior in activity to Ni3N NS/Ti and most reported non-precious metal catalysts under benign conditions. It also performs efficiently for the hydrogen evolution reaction requiring an overpotential of 265 mV for 10 mA cm−2 and its two-electrode electrolyser affords 10 mA cm−2 at a cell voltage of 1.95 V in 0.5 M K–Bi at 25 °C.
Co-reporter:Chun Tang, Zhi Liang Zhao, Jie Chen, Bo Li, Liang Chen, Chang Ming Li
Electrochimica Acta 2017 Volume 248(Volume 248) pp:
Publication Date(Web):10 September 2017
DOI:10.1016/j.electacta.2017.06.159
The direct urea fuel cell holds great promise for energy-sustainable developments and mitigating water contamination but it still faces a great challenge to overcome the sluggish kinetics of the urea oxidation reaction (UOR). In this work, we report Se-Ni(OH)2-shelled vertically oriented NiSe nanowires on a Ni foam as an electrocatalyst toward UOR, showing a low potential of 0.366 V vs. SCE to drive 100 mA cm−2 in alkaline solution and out-performing all the reported non-noble-metal UOR catalysts up to date. Experimental results and theoretical calculation reveal that the vertically and distantly arranged nanowires with highly porous structure produce high mass transport paths for urea to fully access the reaction sites, the NiSe core offers high conductivity for fast electron transport, and the Se-Ni(OH)2 shell provides large amount of active catalytic sites while lowering the CO2 adsorption/desorption barrier than Ni(OH)2 for fast reaction kinetics. This work opens up an exciting new direction to design electrocatalysts for high performance fuel cells and other energy applications.
Co-reporter:Ruixiang Ge;Min Ma;Xiang Ren;Fengli Qu;Zhiang Liu;Gu Du;Abdullah M. Asiri;Baozhan Zheng;Xuping Sun
Chemical Communications 2017 vol. 53(Issue 55) pp:7812-7815
Publication Date(Web):2017/07/06
DOI:10.1039/C7CC03146G
It is attractive but still remains challenging to develop efficient water oxidation electrocatalysts working in a carbonate (Ci) electrolyte. In this communication, we report that a Ni–Co–Ci layer can be developed on a NiCo2O4 nanowire array supported on carbon cloth (NiCo2O4/CC) via electrochemical surface derivation of NiCo2O4. The resulting NiCo2O4@Ni–Co–Ci core–shell nanowire array on carbon cloth (NiCo2O4@Ni–Co–Ci/CC) exhibits high activity toward water oxidation in 1.0 M KHCO3 (K–Ci, pH = 8.3) with the overpotential requirement of 309 mV to drive 10 mA cm−2. NiCo2O4@Ni–Co–Ci/CC also shows long-term electrochemical stability for 20 h and a high turnover frequency of 0.464 mol O2 s−1 at an overpotential of 600 mV.
Co-reporter:Weiyi Wang;Lin Yang;Fengli Qu;Zhiang Liu;Gu Du;Abdullah M. Asiri;Yadong Yao;Xuping Sun
Journal of Materials Chemistry A 2017 vol. 5(Issue 32) pp:16585-16589
Publication Date(Web):2017/08/15
DOI:10.1039/C7TA05521H
Developing non-noble-metal hydrogen evolution reaction electrocatalysts with high activity is critical for future renewable energy systems. Here we describe the development of a self-supported NiMoS4 nanosheet array on Ti mesh (NiMoS4/Ti) through a facile two-step hydrothermal strategy. As a 3D nanoarray electrode for electrochemical hydrogen evolution, NiMoS4/Ti shows exceptionally high catalytic activity and strong durability in 0.1 M KOH (pH: 13). It needs overpotentials of only 194 and 263 mV to drive geometrical catalytic current densities of 10 and 50 mA cm−2, respectively. Moreover, such a catalyst also demonstrates superior long-term stability with a high turnover frequency of 0.75 mol H2 s−1 at an overpotential of 148 mV. Density functional theory calculations suggest a more favorable hydrogen adsorption free energy on the NiMoS4 surface.
Co-reporter:Rong Zhang;Chun Tang;Rongmei Kong;Gu Du;Abdullah M. Asiri;Xuping Sun
Nanoscale (2009-Present) 2017 vol. 9(Issue 14) pp:4793-4800
Publication Date(Web):2017/04/06
DOI:10.1039/C7NR00740J
The scalable production of hydrogen fuel through electrochemical water reduction needs efficient Earth-abundant electrocatalysts to make the whole water-splitting process more energy efficient. In this Article, we report that an Al-doped CoP nanoarray on carbon cloth (Al-CoP/CC) behaves as a durable hydrogen evolution electrocatalyst with superhigh activity in 0.5 M H2SO4. It demands a pretty low overpotential of 23 mV to drive a geometrical catalytic current density of 10 mA cm−2, outperforming all reported non-precious metal catalysts. Density functional theory calculations reveal that Al-CoP has a more thermo-neutral hydrogen adsorption free energy than CoP. Notably, this Al-CoP/CC is also superior in activity and durability as a bifunctional catalyst for alkaline water electrolysis, and its two-electrode water electrolyser delivers 10 mA cm−2 water-splitting current at a cell voltage of 1.56 V in 1.0 M KOH. This work offers us an attractive cost-effective catalyst electrode in water-splitting devices for large-scale production of hydrogen fuels.
Co-reporter:Xiang Ren;Weiyi Wang;Ruixiang Ge;Shuai Hao;Fengli Qu;Gu Du;Abdullah M. Asiri;Qin Wei;Xuping Sun
Chemical Communications 2017 vol. 53(Issue 64) pp:9000-9003
Publication Date(Web):2017/08/08
DOI:10.1039/C7CC03702C
It is highly attractive to develop efficient hydrogen-evolving electrocatalysts under neutral conditions. In this communication, we report an amorphous FeMoS4 nanorod array on carbon cloth (FeMoS4 NRA/CC) prepared by hydrothermal treatment of an FeOOH nanorod array on carbon cloth (FeOOH NRA/CC) in (NH4)2MoS4 solution. As a 3D electrode for hydrogen evolution electrocatalysis, this FeMoS4 NRA/CC demonstrates superior catalytic activity and strong long-term electrochemical durability in 1.0 M phosphate buffered saline (pH: 7). It needs an overpotential of 204 mV to drive a geometrical current density of 10 mA cm−2, which is 450 mV less than that for FeOOH NRA/CC. Density functional theory calculations suggest that FeMoS4 has a more favourable hydrogen adsorption free energy than FeOOH.
Co-reporter:Feng Zhou;Jingjing Zhou;Xuechao Gao;Chunlong Kong
RSC Advances (2011-Present) 2017 vol. 7(Issue 7) pp:3713-3719
Publication Date(Web):2017/01/09
DOI:10.1039/C6RA25396B
A postsynthetic covalent strategy involving dual-acyl chloride has been developed to introduce uncoordinated carboxyl groups into amine containing metal–organic frameworks (MOFs). The carboxyl group functionalized MOFs have been characterized by various techniques, including X-ray diffraction patterning, scanning electron microscopy, Fourier transform infrared spectroscopy, nuclear magnetic resonance, thermal gravimetric analysis, and gas adsorption. Results clearly indicated uncoordinated carboxyl groups were successfully grafted to the MIL-101(Cr)–NH2 framework. In addition, most of the amine groups (>80%) were grafted with carboxyl groups, which indicates this method is very effective. The thermal stability and adsorption selectivity of CO2/N2 were substantially enhanced, albeit the BET surface areas and total pore volumes were reduced. These observations could be explained by the effect of elimination of macropores in the framework due to the projecting of new functional groups in pore apertures. Here the successful fabrication of a MOF with uncoordinated carboxyl groups provides the possibility of efficiently modifying other MOFs.
Co-reporter:Qi Qi, Sujuan Liu, Xing Li, Chunlong Kong, Zhiyong Guo, Liang Chen
Journal of Solid State Chemistry 2017 Volume 255(Volume 255) pp:
Publication Date(Web):1 November 2017
DOI:10.1016/j.jssc.2017.08.004
•High adsorption and catalytic active with extremely low content of the N-NpC about 10.7%.•The catalytic efficiency of the ZnO@N-NpC composites up to 93% after reusing for 5 cycles and stored for two months.•The introduction of N-NpC effectively promotes the photo-induced electron-hole pairs separation, resulting in higher photocatalytic activity.This report describes the controllable encapsulation of ZnO nanoparticles with N-doped nanoporous carbon (N-NpC) via a simple fabrication and calcination of ZnO@ZIF-8 (zeolitic imidazolate framework). In the fabrication of ZnO@ZIF-8, ZnO was used both as the support and Zn source for the formation of ZIF-8. After calcination under N2 atmosphere, the ZnO@N-NpC core-shell heterostructures were formed and characterized by IR, UV–vis, XRD, XPS, scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As expected, the well-defined ZnO@N-NpC core-shell nanospheres demonstrated distinct photocatalytic activity and adsorption capacity in response to the dye methylene blue (MB) in aqueous solution, and the degradation efficiency of MB is up to 99% under UV irradiation for 20 min after catalysts were reused for 5 cycles and stored for two months. Therefore, it is reasonable to believe that the ZnO@N-NpC core-shell heterostructures are new-type nanomaterials for photodegradation of the organic pollutants from wastewater.ZnO@N-doped nanoporous carbon (ZnO@N-NpC) core-shell heterostructures were obtained by a calcination of ZnO@ZIF-8 strategy, which exhibited high adsorption and photocatalytic efficiency up to 93% after 5 cycles and stored for two months.Download high-res image (296KB)Download full-size image
Co-reporter:Chun Tang, Linfeng Gan, Rong Zhang, Wenbo Lu, Xiue Jiang, Abdullah M. Asiri, Xuping Sun, Jin Wang, and Liang Chen
Nano Letters 2016 Volume 16(Issue 10) pp:6617-6621
Publication Date(Web):September 27, 2016
DOI:10.1021/acs.nanolett.6b03332
Replacement of precious Pt with earth-abundant electrocatalysts for the hydrogen evolution reaction (HER) holds great promise for clean energy devices, but the development of low-cost and durable HER catalysts with Pt-like activity is still a huge challenge. In this communication, we report on the development of self-standing ternary FexCo1–xP nanowire array on carbon cloth (FexCo1–xP/CC) as a Pt-free HER catalyst with activities being strongly related to Fe substitution ratio. Electrochemical tests show that Fe0.5Co0.5P/CC not only possesses Pt-like activity with the need of overpotential of only 37 mV to drive 10 mA cm–2, outperforming all non-noble-metal HER catalysts reported to date, but demonstrates superior long-term durability in 0.5 M H2SO4. Density functional theory calculations further reveal that Fe substitution of Co in CoP leads to more optimal free energy of hydrogen adsorption to the catalyst surface. This study offers us a promising flexible monolithic catalyst for practical applications.Keywords: CoP; density functional theory; electrocatalyst; hydrogen evolution reaction; Ternary;
Co-reporter:Jianhui Yang;Qiuju Zhang;Gang Wang;Xiaolong Chen
Advanced Science 2016 Volume 3( Issue 4) pp:
Publication Date(Web):
DOI:10.1002/advs.201500314
Co-reporter:Jianhui Yang, Xuepiao Luo, Shaozheng Zhang and Liang Chen
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 18) pp:12914-12919
Publication Date(Web):08 Apr 2016
DOI:10.1039/C6CP00138F
It is well known that two-dimensional Sc2CT2 (where, T = O, OH or F) structures are excellent semiconductors. The results of the present study employing the first principles calculations indicated that the transition metals, including Ti, V, Cr or Mn, can be doped into Sc2AlC, under ScAl-rich conditions, which is the precursor of the Sc2CT2 structures. The resulting TM-doped Sc2C(OH)2 systems are n-type semiconductors, while the TM-doped Sc2CO2 systems are of the p-type. The difference can be attributed to the higher unfilled p orbitals of O than OH. All Cr- and Mn-doped Sc2CT2 structures were found to be magnetic. Those TM-doped Sc2CT2 systems show heterogeneous magnetic and electronic properties, which can be promising for two-dimensional materials in spin electronics applications.
Co-reporter:Yichao Lin, Chunlong Kong and Liang Chen
RSC Advances 2016 vol. 6(Issue 39) pp:32598-32614
Publication Date(Web):24 Mar 2016
DOI:10.1039/C6RA01536K
We present a review on some recent studies on the syntheses, structures and properties of amine-functionalized metal–organic frameworks (MOFs), and highlight the benefits of amino functionality towards potential applications. Owing to the strong interaction between CO2 and basic amino functionalities, amine-functionalized MOFs have attracted much attention mainly for CO2 capture. Besides the most widely used in situ synthesis method, post-modification and physical impregnation methods are developed to prepare amine-functionalized MOFs with extremely high CO2 sorption capacity at low pressures. On the basis of the similar mechanism, amine-functionalized MOF-based membranes, including pure amine-functionalized MOF membranes and mixed matrix membranes, exhibit excellent CO2/H2, CO2/CH4 and CO2/N2 separation performance. Furthermore, amine-functionalized MOFs also demonstrate potential applications in catalysis.
Co-reporter:Junwen Wang, Yichao Lin, Qunfeng Yue, Kai Tao, Chunlong Kong and Liang Chen
RSC Advances 2016 vol. 6(Issue 58) pp:53017-53024
Publication Date(Web):25 May 2016
DOI:10.1039/C6RA09472D
N-doped porous carbon has a wide range of applications in many fields, such as gas adsorption and super-capacitor, which has stimulated active research for developing efficient strategies to fabricate N-rich porous carbon. In the present study, a series of N-rich porous carbons are derived from polyamine-incorporated metal–organic framework materials (MOFs). Results show that the N content of as-prepared porous carbon is greatly increased by loading polyethyleneimine (PEI) into ZIF-70 frameworks because the loaded PEI can adsorb onto the pore walls of the ZIF-70 and simultaneously supply carbon and N sources during the carbonization process. As a result, the surface area of the as-prepared sample is also increased. In addition, the N content of the porous carbon can be tuned by PEI loadings and carbonization temperature. The as-prepared N-rich porous carbons exhibit greatly enhanced CO2-selective adsorption capacity compared to ZIF-70 and porous carbon derived from ZIF-70. Here the CO2 capture capacity of the as-prepared N-rich porous increases with increasing N content due to considerable interaction affinity between doped N and CO2 molecules. Thus, the as-prepared porous carbon with N content of 11.08 wt% displays high CO2 uptake of 4.86 mmol g−1 at 0 °C and 1 bar, albeit it has a moderate surface area of 652 m2 g−1. Moreover, the N-rich porous carbon clearly shows an excellent separation performance for CO2-over-N2 and CO2-over-CH4. Overall, polyamine-incorporated MOFs are an efficient strategy for controllable fabrication of N-rich porous carbon. The resulting products display a high CO2-selective capture performance. It should be noted that, to achieve the optimal CO2 capture ability, a comprehensive optimization of the polyamine-MOFs-derived porous carbon should be performed.
Co-reporter:Yan Jin, Chongchong Zhao, Zixu Sun, Yichao Lin, Liang Chen, Deyu Wang and Cai Shen
RSC Advances 2016 vol. 6(Issue 36) pp:30763-30768
Publication Date(Web):24 Mar 2016
DOI:10.1039/C6RA01645F
The use of metal organic frameworks (MOFs) as new promising electrode materials in lithium-ion batteries (LIBs) has attracted significant attention. However, the low electrical conductivity of MOFs has resulted in the poor cycle performance of LIBs. Here, we report a facile synthesis route of Fe-MOF/reduced graphene oxide (RGO) composites using a solvothermal method. When used as anode materials for LIBs, the synthesized Fe-MOF/RGO (5%) composite shows superior Li storage with a reversible capacity of 1010.3 mA h g−1 after 200 cycles and an excellent rate performance. The improved electrochemical performance may be attributed to the synergistic effect of MOFs with high theoretical capacities and RGO with high electrical conductivity.
Co-reporter:Lihong Wei, Baihai Li, Qiuju Zhang, Liang Chen, and Xiao Cheng Zeng
The Journal of Physical Chemistry C 2016 Volume 120(Issue 47) pp:26908-26914
Publication Date(Web):November 2, 2016
DOI:10.1021/acs.jpcc.6b09175
Effects of metal hybridization on the electronic properties of the metal–organic framework (MOF) Fe-MOF-74 with different Ni content are investigated using the first-principles method with the Heyd–Scuseria–Ernzerhof (HSE06) functional. The hybrid Ni–Fe-MOF-74, named as the monopolar magnetic semiconductor (MMS), is a new type of porous polarization material that can be easily converted to a half-metal. On the basis of our investigation of the effects of Ni content and the hybrid node arrangement on the band gap of the MOF, we found that the interchain Ni–Fe–Fe arrangement with a Ni content of 33–50% is likely the most suitable configuration due to the narrowing of the spin-down band gap to ∼0.62–0.91 eV from 1.38 eV of the pristine Fe-MOF-74. Hybrid nodes can provide an effective way to narrow the electronic band gap of MOFs and to allow easy conversion of MOFs to polarization materials. Our computation also suggests that the pure Ni-MOF-74, with a band gap of 2.10 eV, can serve as a good visible-light photocatalyst.
Co-reporter:Qiuju Zhang, Minggang Ju, Liang Chen, and Xiao Cheng Zeng
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 17) pp:3395-3400
Publication Date(Web):August 14, 2016
DOI:10.1021/acs.jpclett.6b01507
Two-dimensional (2D) monolayer nanomaterials can be exploited as the thinnest membrane with distinct differential sieving properties for proton isotopes. Motivated from the experimental evidence of differential sieving proton isotopes through graphene and hexagonal boron nitrate (h-BN) monolayer, we compute the kinetic barrier of isotope H+ and D+ permeation through model graphene and h-BN fragments at the MP2/6-31++G(d,p) level of theory. On the basis of the ratio of tunneling reaction rate constant, the isotope separation ratio of H+/D+ and H+/T+ is predicted to be ∼12 and 37, respectively. The tunneling reaction rate constant can be estimated from the zero-point-energy computed at the transition state for the proton isotope permeation though the 2D model systems. We show that the presence of Stone–Wales (55–77) defect in the model graphene fragment can significantly lower the proton permeation barrier by 0.55 eV. With the defect, the ratio of tunneling reaction rate constant of H+/D+ is increased to ∼25. In addition to model graphene and h-BN, we have examined proton permeation capability of α-boron monolayer. We compute the tunneling reaction pathway for H+ through α-boron monolayer using both the climbing nudged elastic band (c-NEB) method and the scanning-path method. Both methods suggest that α-boron monolayer entails a relatively low barrier of ∼0.20 eV for H+ permeation, much lower than that of the model graphene and h-BN fragments. Our studies provide molecular-level insights into the differential permeation of proton isotopes through 2D materials. The methods can be extended to examine isotope separation capability of other 2D materials as well.
Co-reporter:Peipei Zhang, Yibo Hu, Baihai Li, Qiuju Zhang, Chen Zhou, Hongbo Yu, Xuejun Zhang, Liang Chen, Bryan Eichhorn, and Shenghu Zhou
ACS Catalysis 2015 Volume 5(Issue 2) pp:1335
Publication Date(Web):January 16, 2015
DOI:10.1021/cs501612g
This study investigates the structural stability of small Pd@Pt core@shell octahedral nanoparticles (NPs) and their shell thickness dependent catalytic performance for p-chloronitrobenzene hydrogenation with H2. The 6–8 nm Pd@Pt octahedral NPs are prepared by a sequential reduction method, and the characterization results confirm that Pd@Pt octahedral NPs with one to four atomic Pt layers can be controllably synthesized. The Pd@Pt octahedral NPs with one atomic Pt layer demonstrate excellent structural stability with the maintenance of core–shell structures as well as high catalytic stability during cycle to cycle catalytic p-chloronitrobenzene hydrogenation reactions. The alumina-supported Pd@Pt octahedral NPs illustrate a superior catalytic performance relative to individual Pt and Pd and their physical mixtures. Theoretical calculations by density functional theory suggest that the unexpected structural stability for Pd@Pt octahedral NPs with thin Pt shells and their corresponding catalytic stability during hydrogenation reactions can be ascribed to the strong binding between Pt surfaces and reactants/products in catalytic reactions. The enhanced catalytic performance of Pd@Pt octahedral NPs possibly originates from the core–shell interaction, which adjusts the electronic state of surface Pt atoms to be suitable for selective p-chloronitrobenzene hydrogenation.Keywords: core−shell; hydrogenation; nanocatalysis; p-chloronitrobenzene; Pd@Pt
Co-reporter:Yichao Lin, Qiuju Zhang, Chongchong Zhao, Huailong Li, Chunlong Kong, Cai Shen and Liang Chen
Chemical Communications 2015 vol. 51(Issue 4) pp:697-699
Publication Date(Web):11 Nov 2014
DOI:10.1039/C4CC07149B
We present a designed synthesis of a functionalized metal–organic framework with hydrophobic and polar functionalities, which exhibits remarkable thermal and chemical stability. The functionality and porosity make it a promising candidate for the electrode material in Li-ion batteries.
Co-reporter:Yichao Lin, Hao Lin, Haimin Wang, Yange Suo, Baihai Li, Chunlong Kong and Liang Chen
Journal of Materials Chemistry A 2014 vol. 2(Issue 35) pp:14658-14665
Publication Date(Web):30 Jun 2014
DOI:10.1039/C4TA01174K
The global climate change induced by greenhouse gases has stimulated active research for developing efficient strategies to mitigate CO2 emission. In the present study, we prepared a series of polyamine/metal–organic framework (MOF) composites as highly selective CO2 adsorbents from a CO2/N2 mixture, which is relevant to CO2 capture in flue gas. We show that loading polyethyleneimine (PEI) into MIL-101(Cr) frameworks can significantly enhance the selective CO2 adsorption capacity at low pressure and ambient temperature. Further, the comparative study reveals that both the particle size of the MOF and the molecular-weight of PEI play an important role in the CO2 capture ability. Regarding the particle size, smaller MIL-101(Cr) particles can facilitate the loading of PEI into the inner pores and result in lower surface area/pore volume. Thus, the resulting PEI/MIL-101(Cr) composites possess lower CO2 adsorption capacity, but are compensated by higher selectivity of CO2 over N2. On the other hand, lower molecular-weight linear PEI could readily diffuse into the inner pores and effectively block the N2 adsorption. As a result, the as-prepared A-PEI-300 sample in this work exhibits an excellent CO2 uptake of 3.6 mmol g−1 and ultrahigh CO2/N2 selectivity at 0.15 bar and 25 °C. In contrast, the higher molecular-weight branched PEI is advantageous at elevated temperature, since the composites can retain high CO2 adsorption capacity owing to the large amount of primary amine groups. Overall, polyamine/MOF composites are shown to be good candidate adsorbents for CO2 capture from flue gas. To achieve the optimal CO2 capture ability, comprehensive optimization of the polyamine and MOF structures should be performed.
Co-reporter:Kai Tao, Lujie Cao, Yichao Lin, Chunlong Kong and Liang Chen
Journal of Materials Chemistry A 2013 vol. 1(Issue 42) pp:13046-13049
Publication Date(Web):10 Sep 2013
DOI:10.1039/C3TA13371K
A hollow ceramic fiber supported ZIF-8 membrane has been prepared by a hot dip-coating seeding method followed by secondary growth. The obtained membrane exhibits excellent H2 permselectivity.
Co-reporter:Lujie Cao, Kai Tao, Aisheng Huang, Chunlong Kong and Liang Chen
Chemical Communications 2013 vol. 49(Issue 76) pp:8513-8515
Publication Date(Web):30 Jul 2013
DOI:10.1039/C3CC44530E
A thin and compact mixed matrix membrane containing CAU-1-NH2 and the poly(methyl methacrylate) polymer has been originally synthesized. The as-prepared membrane exhibits high permeability of H2 and excellent H2/CO2 selectivity.
Co-reporter:Qiuju Yan, Yichao Lin, Chunlong Kong and Liang Chen
Chemical Communications 2013 vol. 49(Issue 61) pp:6873-6875
Publication Date(Web):10 Jun 2013
DOI:10.1039/C3CC43352H
Solid porous dual amine-decorated metal–organic framework (MOF) adsorbents with tunable porosity have been prepared. The adsorbents exhibit remarkable CO2/CH4 selectivity and CO2 adsorption capacity at low pressures.
Co-reporter:Qiuju Zhang, Baihai Li, and Liang Chen
Inorganic Chemistry 2013 Volume 52(Issue 16) pp:9356-9362
Publication Date(Web):August 6, 2013
DOI:10.1021/ic400927m
A clear understanding of the origin of magnetism in metal–organic frameworks (MOFs) would provide useful insight for tuning the electromagnetic properties of MOFs and finding new applications. In the present study, first-principles calculations show that the open paramagnetic metal sites in three-dimensional porous magnets M-MOF-74 (M = Ni, Co, Fe, Mn) favor high-spin electronic arrangement. Fe- and Co-MOF-74 exhibit ferromagnetic (FM) features and significantly distinct energy gaps between spin-up and spin-down channels in metastable states. After replacement of the Co center with a Ni ion, the FM feature was exhibited for the stable state since the “extra” valence electron was filled in the spin-down 3d bands to shift the Fermi level to higher energy. In contrast, after removal of one valence electron (i.e., replacement of the Fe center with Mn atoms), the energy gap was significantly enlarged and an antiferromagnetic (AFM) feature will be discerned.
Co-reporter:Haiyan Fan, Haiping Xia, Chunlong Kong, Liang Chen
International Journal of Hydrogen Energy 2013 Volume 38(Issue 25) pp:10795-10801
Publication Date(Web):21 August 2013
DOI:10.1016/j.ijhydene.2013.02.040
A thin amine-functionalized MIL-53 membrane with high permeability of hydrogen was successfully prepared on a porous α-Al2O3 support by using the secondary growth method. Seeded α-Al2O3 supports were prepared by a dip-coating technique. In contrast, a discontinuous membrane was obtained by using unseeded support under the same synthesis conditions, implying that the seeds play the key role in the formation of compact membranes. The resulting compact membranes were measured by X-ray diffraction (XRD), scanning electron microscopy (SEM) and single gas permeation testing. Results showed that the thickness of the as-prepared membrane was around 2 ∼ 4 μm. Hydrogen permeance of the as-prepared membrane reached a remarkable value of 1.5 × 10−5 mol m−2 s−1·Pa−1 at room temperature under a 0.1 MPa pressure drop. The ideal H2/CO2 selectivity was found to be 4.4. In addition, the influence of seeding solution on the membrane performance was investigated. We found that the membrane permeance decreased and the ideal selectivity increased when the seeding solution content was increased.Highlights► A thin functionalized MIL-53 membrane was prepared on a porous α-Al2O3 support. ► The seeded α-Al2O3 support plays the key role in the formation of compact membranes. ► Hydrogen permeance of the membrane reached a remarkable value at room temperature. ► The ideal H2/CO2 selectivity of the as-prepared membrane was found to be 4.4.
Co-reporter:Yumin Leng, Yonglong, Li, An Gong, Zheyu Shen, Liang Chen, and Aiguo Wu
Langmuir 2013 Volume 29(Issue 25) pp:7591-7599
Publication Date(Web):May 31, 2013
DOI:10.1021/la400909b
A new kind of analytical reagent, hexadecyl trimethyl ammonium bromide (CTAB), and dithizone product-modified gold nanoparticle dispersion, is developed for colorimetric response to 10 types of heavy metal ions (Mn+), including Cr(VI), Cr3+, Mn2+, Co2+, Ni2+, Cu2+, Zn2+, Cd2+, Hg2+, and Pb2+. The color change of the modified gold nanoparticle dispersion is instantaneous and distinct for Mn2+, Co2+, Ni2+, Cu2+, Zn2+, Cd2+, Hg2+, and Pb2+. The color change results from the multiple reasons, such as electronic transitions, cation−π interactions, formation of coordination bonds, and Mn+-induced aggregation of gold nanoparticles (AuNPs). The different combining capacity of heavy metal ions to modifiers results in the different broadening and red-shifting of the plasmon peak of modified AuNPs. In addition, Cr(VI), Cu2+, Co2+, Ni2+, and Mn2+ cause the new UV–vis absorption peaks in the region of 360–460 nm. The interactions between the modifiers and AuNPs, and between the modifiers and Mn+, are investigated by using Fourier transform infrared spectroscopy and X-ray photoelectron spectroscopy. The results confirm that AuNPs are modified by CTAB and dithizone products through electrostatic interactions and Au–S bonds, respectively, and the Mn+–N bonds form between Mn+ and dithizone products. Furthermore, the experimental and density functional theory calculated IR spectra prove that dithizone reacts with NaOH to produce C6H5O– and [SCH2N4]2-. The validation of this method is carried out by analysis of heavy metal ions in tap water.
Co-reporter:Qiuju Yan;Yichao Lin;Dr. Pengyan Wu;Li Zhao;Lujie Cao; Luming Peng;Assoc. Chunlong Kong; Liang Chen
ChemPlusChem 2013 Volume 78( Issue 1) pp:86-91
Publication Date(Web):
DOI:10.1002/cplu.201200270
Abstract
In metal–organic framework (MOF) chemistry, polar functionalities greatly affect the gas adsorption properties. However, synthesis of MOFs with desired functionality is very challenging because many chemical functionalities cannot be achieved under the conditions for MOF assembly. Herein, a facile synthesis of new functionalized two-dimensional MOFs with preferential CO2 capture is presented, which uses two successive synthesis steps: 1) rational design and template-free synthesis of the parent MOF with designated pendant amino groups and 2) postsynthetic modification of the active amino groups with acetic acid and trimesoyl chloride functionalities. The only variation in structure arises from the functional groups of these materials. Experimental results demonstrate that the three 2D layered MOFs have remarkable thermal stability and moisture resistance, which are particularly advantageous for practical CO2 capture. Although their surface areas are moderate (270–340 m2 g−1), they still have excellent CO2 adsorption capacity (up to 2.9 mmol g−1 at 1 bar and 273 K) comparable to that of previously reported MOFs with much higher surface areas. Based on first-principles calculations, it is shown that the acidic carbonyl functionalities in addition to the amino groups are also favorable to bind CO2 molecules. The adsorption sites generated from polar functionalities are key factors leading to high CO2 uptake.
Co-reporter:Qiuju Zhang, Lujie Cao, Baihai Li and Liang Chen
Chemical Science 2012 vol. 3(Issue 9) pp:2708-2715
Publication Date(Web):27 Jun 2012
DOI:10.1039/C2SC20521A
A metal–organic framework (MOF)-based catalyst W–Cu–BTC is designed by hybridizing highly active W ions into Cu3(BTC)2(H2O)3 (also known as Cu–BTC or HKUST-1, BTC = 1,3,5-tricarboxylate benzene) frameworks based on density functional (DFT) calculations. We show that the hybrid W–Cu node plays a pivotal role in activating CO2 according to frontier molecular orbital theory. In contrast to the Lewis-acid nature of open metal sites in most MOFs, the exposed W ion in W–Cu–BTC is identified as a Lewis-base site, evidenced by the substantial electron donation from W ion to CO2. Kinetically, the linear CO2 molecule can be readily bent by forming a CO2–W complex after overcoming a negligible activation barrier of 0.09 eV. In addition, we present calculated infrared spectra (IR) and X-Ray spectra (XPS) for reference in future experimental studies.
Co-reporter:Yichao Lin, Chunlong Kong and Liang Chen
RSC Advances 2012 vol. 2(Issue 16) pp:6417-6419
Publication Date(Web):13 Jun 2012
DOI:10.1039/C2RA20641B
A pure amine-functionalized MIL-101(Cr) has been synthesized for the first time by a simple method. The as-prepared nanoparticles are around 50 nm. In addition, the resulting amine-functionalized MIL-101(Cr) displayed excellent CO2 adsorption capacity, up to 15 mmol g−1 at 16 °C.
Co-reporter:Tongyu Wang, Qiuju Zhang, Baihai Li, Hong Chen, Liang Chen
International Journal of Hydrogen Energy 2012 Volume 37(Issue 6) pp:5081-5089
Publication Date(Web):March 2012
DOI:10.1016/j.ijhydene.2011.12.065
The direct doping of small Pd cluster on IRMOF-1 and its influence on H2 adsorption were investigated using periodic density functional methods. Our calculations indicate that the Pd cluster prefers to be deposited on the linker. The doped Pd cluster can not only play the important role for dissociating H2 molecules, but also enhance the H interaction with IRMOF-1 by altering its electronic structures. As a result, hydrogen spillover in IRMOF-1 by directly doping Pd catalysts is thermodynamically feasible and the odd numbered H bottlenecks on undoped IRMOF-1 are eliminated. In addition, we discuss a working mechanism for releasing H2 from the chemisorbed states.Highlights▶ Pd clusters prefer to be doped on the linker of IRMOF-1. ▶ The doped Pd clusters enhance H interactions with IRMOF-1 by altering its electronic structures. ▶ The odd numbered H bottlenecks on undoped IRMOF-1 are eliminated.
Co-reporter:Houyuan Wang, Shihao Wei, Qiuju Zhang, Liang Chen
Computational and Theoretical Chemistry 2012 Volume 999() pp:162-168
Publication Date(Web):1 November 2012
DOI:10.1016/j.comptc.2012.08.032
To elucidate the peak shifting of C-1s in X-ray photoelectron spectrum (XPS) of perchloroethylene (PCE) adsorption on Si(1 0 0), the sequential adjacent Si dimer dechlorination mechanism was proposed based on first principles calculations. The highly-symmetric Cl atoms of PCE induce three possible initial di-dechlorination processes occurring on intra, inter-dimer and iso intra-dimer, respectively, to yield three tetra-σ states. These tetra-σ states are identified to coexist at room temperature (RT) due to the relatively low reaction barriers (<0.59 eV). However, their further di-dechlorination to form intra and inter-dimer hexa-σ states requires much higher activation barriers (>1.08 eV), which leads to hexa-σ states only exist at elevated temperatures although they are found to be the most stable in terms of energetics. The calculated ionization energies (IEs) of C-1s core electron and vibrational frequencies of various potential adspecies are well consistent with the experimental data observed by XPS and vibrational electron energy loss spectroscopy (EELS), which further corroborates the sequential dechlorination processes of PCE on Si(1 0 0).Graphical abstractThe adjacent Si dimer dechlorination processes of PCE/Si(1 0 0) at RT.Highlights► PCE/Si(1 0 0) undergoes sequential di-dechlorination with temperature evolution. ► Three tetra-σ adspecies coexist at room temperature after the first di-dechlorination. ► Two hexa-σ states are produced by further di-dechlorination at high temperature. ► The calculated IEs of C-1s and vibrational frequencies well interpret XPS and EELS.
Co-reporter:Yumin Leng, Fuqiang Zhang, Yujie Zhang, Xiaoqin Fu, Yanbo Weng, Liang Chen, Aiguo Wu
Talanta 2012 Volume 94() pp:271-277
Publication Date(Web):30 May 2012
DOI:10.1016/j.talanta.2012.03.039
We previously reported a colorimetric assay method for Co2+ based on the thioglycolic acid (TGA) functionalized hexadecyl trimethyl ammonium bromide (CTAB) modified Au NPs. However, the detection limit of 3 × 10−7 M was still higher than that of the sanitary standard for drinking water (6.8 × 10−8 M). In addition, the interactions between the modifier and Au NPs, and between the modifier-Au NPs and Co2+ remain to be clarified and confirmed. Thus, in the present study, the modified Au NPs solution was dialyzed and its detection limit was optimized to be 5 × 10−10 M. The interactions between the modifier and Au NPs, and between the modifier-Au NPs and Co2+ were investigated in both experimental characterizations and theoretical calculations, consistently confirming that the Au NPs were modified by the negatively charged anions of [SCH2CO2]2− through Au–S bonds and Co2+ was recognized by the modifier-Au NPs through CoO chelate bonds. The results of X-ray photoelectron spectroscopy (XPS) suggest that there were no chemical bonds formed between CTAB and Co2+. Moreover, the colorimetric assay of Co2+ using the modified Au NPs has been proved to be a rapid, very sensitive and highly selective method. The validation of the method was carried out by analysis of a certified reference material, GSBZ 50030-94.Highlights► A highly rapid and sensitive colorimetric method for Co2+ detection was developed. ► The detection time was within 1 min. ► The detection limit was about 5 × 10−10 M. ► The involved interactions were confirmed by experiments and simulations.
Co-reporter:Jianhui Yang, Bo Li, Qiuju Zhang, Wai-leung Yim, and Liang Chen
The Journal of Physical Chemistry C 2012 Volume 116(Issue 20) pp:11189-11194
Publication Date(Web):May 6, 2012
DOI:10.1021/jp302865z
Oxygen activation on one-dimensional (1D) gold nanotube T(6,0) and nanowire W6-1 was investigated using first principles density functional methods (DFT). It is found that the oxygen adsorption strength on T(6,0) is unexpectedly weak, whereas W6-1 with inner Au–Au bonds is more active for O2 adsorption. To further promote the catalyzed oxygen activation, we proposed and evaluated two working strategies, including imposing a small strain along the axial direction and doping alien transition metal (TM) atoms. Specifically, 4% compression or doping Pt atoms can decrease the O2 dissociation barriers from 0.85 eV to a more desired 0.6 eV. The analysis of electronic structures and charge density difference revealed that the inner Au–TM bond can decrease the filling degree of the Au-dz2 orbital and, thus, improve the oxygen activity.
Co-reporter:Qiuju Zhang, Bo Li, Qinghong Yuan, Baihai Li, Zhifeng Liu and Liang Chen
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 15) pp:7121-7128
Publication Date(Web):14 Mar 2011
DOI:10.1039/C0CP01506G
The dechlorination processes of isomers trans and iso-dichloroethylene (iso-DCE) on Si(100)-2×1 were investigated from first principles, to ascertain the isomeric effect on the adjacent Si dimer di-dechlorination of DCE on Si(100)-2×1. By comparing the feasible adspecies and their reaction barriers between trans and cis-DCE on Si(100)-2×1, we found that the isomeric effect of trans-DCE is negligible, which explained the similar C 1s peak locations in the X-ray photoelectron spectroscopy (XPS) experiment. In contrast, iso-DCE undergoes a more complicated reaction process, although the adjacent Si dimer di-dechlorination is still the dominant mechanism. Among the initial competitive reactions involving intra-, inter-cycloaddition and single C–Cl cleavage, the barrierless intra-cycloaddition is the most favorable reaction and precludes the single dechlorination that yields the mono-σ structure. Subsequent di-dechlorination undergoes a three-step reaction to yield the final product intra-dimer tetra-σ. In addition, the ionization energies of C 1s and Cl 2s electrons were calculated for the tentative assignment of the peaks observed in XPS.
Co-reporter:Tongyu Wang, Baihai Li, Jianhui Yang, Hong Chen and Liang Chen
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 15) pp:7112-7120
Publication Date(Web):16 Mar 2011
DOI:10.1039/C0CP02007A
The formation of Pd/Au surfaces and their catalytic performance toward oxygen dissociation were investigated using periodic density functional methods. We show that Pd can readily incorporate into the second layer of Au(100) and Au(111) substrates with the assistance of Au vacancies. Pd/Au(100) exhibits better catalytic activity toward oxygen dissociation than Pd/Au(111). Specifically, the sub-layer Pd atoms of Pd/Au(100) can promote the oxygen dissociation and stabilize the surface structure after adsorbing oxygen atoms. On the contrary, the sub-layer Pd atoms of Pd/Au(111) slightly hinder the oxygen dissociation.
Co-reporter:Chenggang Zhou, Lujie Cao, Shihao Wei, Qiuju Zhang, Liang Chen
Computational and Theoretical Chemistry 2011 Volume 976(1–3) pp:153-160
Publication Date(Web):1 December 2011
DOI:10.1016/j.comptc.2011.08.018
We examined the adsorption of selected guest molecules on the open Cu sites with different charge states in CuBTC by means of density functional methods. Our calculations revealed that CO and O2 are chemisorbed on the open Cu site, whereas CO2, H2 and CH4 are mainly physisorbed. As an exception, the adsorption nature of N2 is dependent on the charge state of the open Cu ion. The reduced positively charged Cu ion can significantly enhance binding strength of CO, O2 and N2 because of the chemisorptive nature. On the other hand, losing electrons from the open Cu ion can slightly improve the physisorption strength through electrostatic interactions. Therefore, it may be possible to enhance gas adsorption and control selective separation for specific gas mixtures by appropriately tuning charge state of the exposed metal in metal organic frameworks.Graphical abstractTuning charge state of the open Cu ion of dehydrated Cu3(BTC)2 to enhance the adsorption of the gas molecules.Highlights► Charge variation of the open Cu in CuBTC influences the gas adsorption behaviors. ► The binding strengths of CO, O2 and N2 are enhanced in negatively-charged CuBTC. ► Their binding nature exhibits chemisorptive. ► Positively-charged CuBTC slightly improves the adsorption strength of CO2.
Co-reporter:Baihai Li, Qiuju Zhang, Liang Chen, Ping Cui and Xiaoqing Pan
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 28) pp:7848-7855
Publication Date(Web):24 May 2010
DOI:10.1039/B925764K
We present a theoretical understanding of carbon deposition and diffusion in FCC and HCP cobalt from first principles. We found that the deposited carbon atom can readily penetrate into the first sub-layer of Co substrates, while further diffusion into deeper interstices seems unfeasible. In the presence of cobalt vacancy, the carbon diffusion can be greatly promoted and possibly leads to the carburization of cobalt catalysts. The infiltrated carbon atoms have a pronounced influence on the catalytic activity toward CO adsorption and dissociation. Compared to the clean cobalt surfaces, the C–O bond is less weakened on the carburized cobalt due to the depletion of Co-d electrons. As a result, the activation barrier for CO dissociation is substantially increased. We suggest that the carburization is another important cause to the deactivation of Co-based catalysts in addition to the site blockage by the surface carbon deposition.
Co-reporter:Baihai Li, Wai-Leung Yim, Qiuju Zhang and Liang Chen
The Journal of Physical Chemistry C 2010 Volume 114(Issue 7) pp:3052-3058
Publication Date(Web):January 28, 2010
DOI:10.1021/jp9098396
Hydrogen chemisorption and diffusion on Pt and Pd decorated MoO3(010) surfaces were examined using periodic density functional methods. The deposition of Pt and Pd on MoO3 was first carefully investigated. The strong metal−support interactions were found to greatly reduce the catalytic activity of Pt and Pd atoms anchored at their most favorable binding sites. On the other hand, the energies and activation barriers along selected diffusion pathways indicate that hydrogen dissociation and diffusion on the supported Pt5 and Pd5 clusters are feasible, whereas Pt clusters exhibit better catalytic activity than Pd clusters. Subsequently, the dissociated hydrogen atoms tend to directly diffuse onto the sublayer oxygen atoms instead of the surface oxygen atoms.
Co-reporter:Qiuju Zhang, Zhifeng Liu, Baihai Li and Liang Chen
The Journal of Physical Chemistry C 2009 Volume 113(Issue 52) pp:21797-21804
Publication Date(Web):December 10, 2009
DOI:10.1021/jp9077954
The dehalogenation mechanisms of cis-dichloroethylene (cis-DCE) and cis-dibromoethylene (cis-DBE) adsorption on Si(100)2 × 1 are explored by using a first principles method to understand the halogen substitution effects. Instead of the two C−X (X = Cl, Br) bonds cleaved on a single broken Si dimer to produce the vinylene di-σ product, a newly identified adjacent Si dimer didechlorination mode is found to be more favorable. This double dechlorination is a two-step reaction, including the initial cycloaddition and the following didechlorination, with activation barriers of no more than 0.40 eV. The overall reaction yields two new adspecies, named as the intra- and interdimer tetra-σ. The mono-σ adspecies derived from one C−Cl cleavage is a minor product, although it is thermally less favorable than the intra- and interdimer tetra-σ states. With thermal evolution, its conversion to the final product (interdimer tetra-σ) is investigated, which accounts for the C 1s photoelectron peaks shifting from 285.6 to 283.9 eV at higher temperature observed in the X-ray photoelectron spectroscopy (XPS) experiments. The core level shifts of the C 1s electron in various potential adspecies are also calculated to compare with the photoelectron peaks obtained in the XPS spectra.
Co-reporter:Liang Chen and Bei Chen, Chenggang Zhou and Jinping Wu, Robert C. Forrey, Hansong Cheng
The Journal of Physical Chemistry C 2008 Volume 112(Issue 36) pp:13937-13942
Publication Date(Web):2017-2-22
DOI:10.1021/jp803504k
We present a systematic study on the reactivity of a Pt6 cluster toward H2 dissociative chemisorption in the presence of CO molecules using density functional theory (DFT). The sequential adsorption and threshold desorption energies of H at varying CO coverage were identified. It was found that the main influence of CO molecules is to block the available active surface sites for H2 dissociative chemisorption. In addition, our population and density of states analysis indicate that the poisoning effect is partially due to the loss of Pt(5d) electrons upon CO adsorption. The hydrogenation of CO is found to be endothermic.
Co-reporter:Xuechao Gao, Guozhao Ji, Li Peng, Xuehong Gu, Liang Chen
Journal of Membrane Science (1 June 2017) Volume 531() pp:36-46
Publication Date(Web):1 June 2017
DOI:10.1016/j.memsci.2017.02.035
Co-reporter:Qiuju Zhang, Lujie Cao, Baihai Li and Liang Chen
Chemical Science (2010-Present) 2012 - vol. 3(Issue 9) pp:NaN2715-2715
Publication Date(Web):2012/06/27
DOI:10.1039/C2SC20521A
A metal–organic framework (MOF)-based catalyst W–Cu–BTC is designed by hybridizing highly active W ions into Cu3(BTC)2(H2O)3 (also known as Cu–BTC or HKUST-1, BTC = 1,3,5-tricarboxylate benzene) frameworks based on density functional (DFT) calculations. We show that the hybrid W–Cu node plays a pivotal role in activating CO2 according to frontier molecular orbital theory. In contrast to the Lewis-acid nature of open metal sites in most MOFs, the exposed W ion in W–Cu–BTC is identified as a Lewis-base site, evidenced by the substantial electron donation from W ion to CO2. Kinetically, the linear CO2 molecule can be readily bent by forming a CO2–W complex after overcoming a negligible activation barrier of 0.09 eV. In addition, we present calculated infrared spectra (IR) and X-Ray spectra (XPS) for reference in future experimental studies.
Co-reporter:Ling Zhang, Lisi Xie, Min Ma, Fengli Qu, Gu Du, Abdullah M. Asiri, Liang Chen and Xuping Sun
Catalysis Science & Technology (2011-Present) 2017 - vol. 7(Issue 13) pp:NaN2694-2694
Publication Date(Web):2017/05/23
DOI:10.1039/C7CY00703E
Designing and developing non-noble-metal electrocatalysts for efficient full water splitting at neutral pH is highly desired but still remains a huge challenge. In this communication, we report the development of homologous Co-based nanowire films, Co2N nanowire film on a Ti mesh (Co2N/TM) and Co-phosphate nanowire film on a Ti mesh (Co–Pi/TM), as complementary catalysts for stable electrochemical hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) in neutral electrolyte. Co2N/TM shows remarkable HER activity with the need of an overpotential of 290 mV to drive 10 mA cm−2 in 1.0 M phosphate-buffered saline (PBS), and Co–Pi/TM is superior in OER activity affording 10 mA cm−2 at overpotentials of only 430 and 300 mV in 0.1 and 1.0 M PBS, respectively. The two-electrode water electrolyzer using Co2N/TM as a cathode and Co–Pi/TM as an anode affords a water-splitting current density of 10 mA cm−2 at a cell voltage of 1.78 V with 100% Faradaic efficiency in 1.0 M PBS, promising the practical uses of such homologous Co-based catalyst materials in technological devices.
Co-reporter:Lisi Xie, Fengli Qu, Zhiang Liu, Xiang Ren, Shuai Hao, Ruixiang Ge, Gu Du, Abdullah M. Asiri, Xuping Sun and Liang Chen
Journal of Materials Chemistry A 2017 - vol. 5(Issue 17) pp:NaN7810-7810
Publication Date(Web):2017/04/10
DOI:10.1039/C7TA02333B
It is of great importance but still remains a key challenge to develop non-noble-metal bifunctional catalysts for efficient full water splitting under mild pH conditions. In this communication, we report the in situ electrochemical development of an ultrathin Ni–Bi layer on a metallic Ni3N nanosheet array supported on a Ti mesh (Ni3N@Ni–Bi NS/Ti) as a durable 3D core/shell structured nanoarray electrocatalyst for water oxidation at near-neutral pH. The Ni3N@Ni–Bi NS/Ti demands overpotentials of 405 and 382 mV to deliver a geometrical catalytic current density of 10 mA cm−2 in 0.1 and 0.5 M K–Bi (pH: 9.2), respectively, superior in activity to Ni3N NS/Ti and most reported non-precious metal catalysts under benign conditions. It also performs efficiently for the hydrogen evolution reaction requiring an overpotential of 265 mV for 10 mA cm−2 and its two-electrode electrolyser affords 10 mA cm−2 at a cell voltage of 1.95 V in 0.5 M K–Bi at 25 °C.
Co-reporter:Qiuju Zhang, Bo Li, Qinghong Yuan, Baihai Li, Zhifeng Liu and Liang Chen
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 15) pp:NaN7128-7128
Publication Date(Web):2011/03/14
DOI:10.1039/C0CP01506G
The dechlorination processes of isomers trans and iso-dichloroethylene (iso-DCE) on Si(100)-2×1 were investigated from first principles, to ascertain the isomeric effect on the adjacent Si dimer di-dechlorination of DCE on Si(100)-2×1. By comparing the feasible adspecies and their reaction barriers between trans and cis-DCE on Si(100)-2×1, we found that the isomeric effect of trans-DCE is negligible, which explained the similar C 1s peak locations in the X-ray photoelectron spectroscopy (XPS) experiment. In contrast, iso-DCE undergoes a more complicated reaction process, although the adjacent Si dimer di-dechlorination is still the dominant mechanism. Among the initial competitive reactions involving intra-, inter-cycloaddition and single C–Cl cleavage, the barrierless intra-cycloaddition is the most favorable reaction and precludes the single dechlorination that yields the mono-σ structure. Subsequent di-dechlorination undergoes a three-step reaction to yield the final product intra-dimer tetra-σ. In addition, the ionization energies of C 1s and Cl 2s electrons were calculated for the tentative assignment of the peaks observed in XPS.
Co-reporter:Yichao Lin, Qiuju Zhang, Chongchong Zhao, Huailong Li, Chunlong Kong, Cai Shen and Liang Chen
Chemical Communications 2015 - vol. 51(Issue 4) pp:NaN699-699
Publication Date(Web):2014/11/11
DOI:10.1039/C4CC07149B
We present a designed synthesis of a functionalized metal–organic framework with hydrophobic and polar functionalities, which exhibits remarkable thermal and chemical stability. The functionality and porosity make it a promising candidate for the electrode material in Li-ion batteries.
Co-reporter:Qiuju Yan, Yichao Lin, Chunlong Kong and Liang Chen
Chemical Communications 2013 - vol. 49(Issue 61) pp:NaN6875-6875
Publication Date(Web):2013/06/10
DOI:10.1039/C3CC43352H
Solid porous dual amine-decorated metal–organic framework (MOF) adsorbents with tunable porosity have been prepared. The adsorbents exhibit remarkable CO2/CH4 selectivity and CO2 adsorption capacity at low pressures.
Co-reporter:Tongyu Wang, Baihai Li, Jianhui Yang, Hong Chen and Liang Chen
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 15) pp:NaN7120-7120
Publication Date(Web):2011/03/16
DOI:10.1039/C0CP02007A
The formation of Pd/Au surfaces and their catalytic performance toward oxygen dissociation were investigated using periodic density functional methods. We show that Pd can readily incorporate into the second layer of Au(100) and Au(111) substrates with the assistance of Au vacancies. Pd/Au(100) exhibits better catalytic activity toward oxygen dissociation than Pd/Au(111). Specifically, the sub-layer Pd atoms of Pd/Au(100) can promote the oxygen dissociation and stabilize the surface structure after adsorbing oxygen atoms. On the contrary, the sub-layer Pd atoms of Pd/Au(111) slightly hinder the oxygen dissociation.
Co-reporter:Lujie Cao, Kai Tao, Aisheng Huang, Chunlong Kong and Liang Chen
Chemical Communications 2013 - vol. 49(Issue 76) pp:NaN8515-8515
Publication Date(Web):2013/07/30
DOI:10.1039/C3CC44530E
A thin and compact mixed matrix membrane containing CAU-1-NH2 and the poly(methyl methacrylate) polymer has been originally synthesized. The as-prepared membrane exhibits high permeability of H2 and excellent H2/CO2 selectivity.
Co-reporter:Kai Tao, Lujie Cao, Yichao Lin, Chunlong Kong and Liang Chen
Journal of Materials Chemistry A 2013 - vol. 1(Issue 42) pp:NaN13049-13049
Publication Date(Web):2013/09/10
DOI:10.1039/C3TA13371K
A hollow ceramic fiber supported ZIF-8 membrane has been prepared by a hot dip-coating seeding method followed by secondary growth. The obtained membrane exhibits excellent H2 permselectivity.
Co-reporter:Ruixiang Ge, Min Ma, Xiang Ren, Fengli Qu, Zhiang Liu, Gu Du, Abdullah M. Asiri, Liang Chen, Baozhan Zheng and Xuping Sun
Chemical Communications 2017 - vol. 53(Issue 55) pp:NaN7815-7815
Publication Date(Web):2017/05/24
DOI:10.1039/C7CC03146G
It is attractive but still remains challenging to develop efficient water oxidation electrocatalysts working in a carbonate (Ci) electrolyte. In this communication, we report that a Ni–Co–Ci layer can be developed on a NiCo2O4 nanowire array supported on carbon cloth (NiCo2O4/CC) via electrochemical surface derivation of NiCo2O4. The resulting NiCo2O4@Ni–Co–Ci core–shell nanowire array on carbon cloth (NiCo2O4@Ni–Co–Ci/CC) exhibits high activity toward water oxidation in 1.0 M KHCO3 (K–Ci, pH = 8.3) with the overpotential requirement of 309 mV to drive 10 mA cm−2. NiCo2O4@Ni–Co–Ci/CC also shows long-term electrochemical stability for 20 h and a high turnover frequency of 0.464 mol O2 s−1 at an overpotential of 600 mV.
Co-reporter:Baihai Li, Qiuju Zhang, Liang Chen, Ping Cui and Xiaoqing Pan
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 28) pp:NaN7855-7855
Publication Date(Web):2010/05/24
DOI:10.1039/B925764K
We present a theoretical understanding of carbon deposition and diffusion in FCC and HCP cobalt from first principles. We found that the deposited carbon atom can readily penetrate into the first sub-layer of Co substrates, while further diffusion into deeper interstices seems unfeasible. In the presence of cobalt vacancy, the carbon diffusion can be greatly promoted and possibly leads to the carburization of cobalt catalysts. The infiltrated carbon atoms have a pronounced influence on the catalytic activity toward CO adsorption and dissociation. Compared to the clean cobalt surfaces, the C–O bond is less weakened on the carburized cobalt due to the depletion of Co-d electrons. As a result, the activation barrier for CO dissociation is substantially increased. We suggest that the carburization is another important cause to the deactivation of Co-based catalysts in addition to the site blockage by the surface carbon deposition.
Co-reporter:Jianhui Yang, Xuepiao Luo, Shaozheng Zhang and Liang Chen
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 18) pp:NaN12919-12919
Publication Date(Web):2016/04/08
DOI:10.1039/C6CP00138F
It is well known that two-dimensional Sc2CT2 (where, T = O, OH or F) structures are excellent semiconductors. The results of the present study employing the first principles calculations indicated that the transition metals, including Ti, V, Cr or Mn, can be doped into Sc2AlC, under ScAl-rich conditions, which is the precursor of the Sc2CT2 structures. The resulting TM-doped Sc2C(OH)2 systems are n-type semiconductors, while the TM-doped Sc2CO2 systems are of the p-type. The difference can be attributed to the higher unfilled p orbitals of O than OH. All Cr- and Mn-doped Sc2CT2 structures were found to be magnetic. Those TM-doped Sc2CT2 systems show heterogeneous magnetic and electronic properties, which can be promising for two-dimensional materials in spin electronics applications.
Co-reporter:Yichao Lin, Hao Lin, Haimin Wang, Yange Suo, Baihai Li, Chunlong Kong and Liang Chen
Journal of Materials Chemistry A 2014 - vol. 2(Issue 35) pp:NaN14665-14665
Publication Date(Web):2014/06/30
DOI:10.1039/C4TA01174K
The global climate change induced by greenhouse gases has stimulated active research for developing efficient strategies to mitigate CO2 emission. In the present study, we prepared a series of polyamine/metal–organic framework (MOF) composites as highly selective CO2 adsorbents from a CO2/N2 mixture, which is relevant to CO2 capture in flue gas. We show that loading polyethyleneimine (PEI) into MIL-101(Cr) frameworks can significantly enhance the selective CO2 adsorption capacity at low pressure and ambient temperature. Further, the comparative study reveals that both the particle size of the MOF and the molecular-weight of PEI play an important role in the CO2 capture ability. Regarding the particle size, smaller MIL-101(Cr) particles can facilitate the loading of PEI into the inner pores and result in lower surface area/pore volume. Thus, the resulting PEI/MIL-101(Cr) composites possess lower CO2 adsorption capacity, but are compensated by higher selectivity of CO2 over N2. On the other hand, lower molecular-weight linear PEI could readily diffuse into the inner pores and effectively block the N2 adsorption. As a result, the as-prepared A-PEI-300 sample in this work exhibits an excellent CO2 uptake of 3.6 mmol g−1 and ultrahigh CO2/N2 selectivity at 0.15 bar and 25 °C. In contrast, the higher molecular-weight branched PEI is advantageous at elevated temperature, since the composites can retain high CO2 adsorption capacity owing to the large amount of primary amine groups. Overall, polyamine/MOF composites are shown to be good candidate adsorbents for CO2 capture from flue gas. To achieve the optimal CO2 capture ability, comprehensive optimization of the polyamine and MOF structures should be performed.




](http://img.cochemist.com/ccimg/27000/26913-06-4.png)
](http://img.cochemist.com/ccimg/27000/26913-06-4_b.png)