Based on 1-amino-4-hydroxy-triptycene, new saturated and unsaturated triptycene-NHC (N-heterocyclic carbene) ligands were synthesized from glyoxal-derived diimines. The respective carbenes were converted into metal complexes [(NHC)MX] (M=Cu, Ag, Au; X=Cl, Br) and [(NHC)MCl(cod)] (M=Rh, Ir; cod=1,5-cyclooctadiene) in good yields. The new azolium salts and metal complexes suffer from limited solubility in common organic solvents. Consequently, the introduction of solubilizing groups (such as 2-ethylhexyl or 1-hexyl by O-alkylation) is essential to render the complexes soluble. The triptycene unit infers special steric properties onto the metal complexes that enable the steric shielding of selected areas close to the metal center. Next, chiral and meso-triptycene based N-heterocyclic carbene ligands were prepared. The key step in the synthesis of the chiral ligand is the Buchwald–Hartwig amination of 1-bromo-4-butoxy-triptycene with (1S,2S)-1,2-diphenyl-1,2-diaminoethane, followed by cyclization to the azolinium salt with HC(OEt)3. The analogous reaction with meso-1,2-diphenyl-1,2-diaminoethane provides the respective meso-azolinium salt. Both the chiral and meso-azolinium salts were converted into metal complexes including [(NHC)AuCl], [(NHC)RhCl(cod)], [(NHC)IrCl(cod)], and [(NHC)PdCl(allyl)]. An in situ prepared chiral copper complex was tested in the enantioselective borylation of α,β-unsaturated esters and found to give an excellent enantiomeric ratio (er close to 90:10).
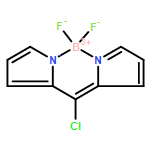
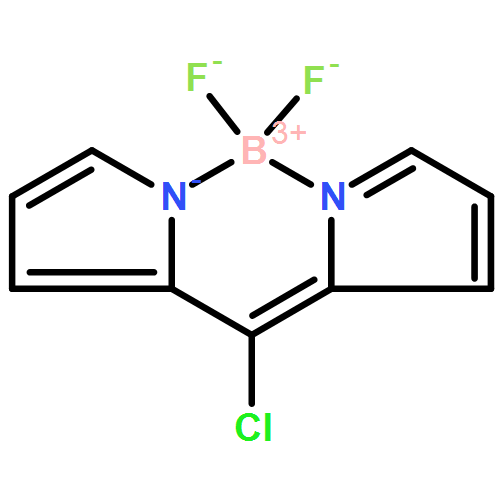
The Pd-catalyzed reactions of 3-chloro-bodipy with R2PH (R=Ph, Cy) provide nonfluorescent bodipy–phosphines 3-PR2–bodipy 3 a (R=Ph) and 3 b (R=Cy; quantum yield Φ<0.001). Metal complexes such as [AgCl(3 b)] and [AuCl(3 b)] were prepared and shown to display much higher fluorescence (Φ=0.073 and 0.096). In the gold complexes, the level of fluorescence was found to be qualitatively correlated with the electron density at gold. Consequently, the fluorescence brightness of [AuCl(3 b)] increases when the chloro ligand is replaced by a weakly coordinating anion, whereas upon formation of the electron-rich complex [Au(SR)(3 b)] the fluorescence is almost quenched. Related reactions of [AuCl(3 b)] with [Ag]ONf)] (Nf= nonaflate) and phenyl acetylenes enable the tracking of initial steps in gold-catalyzed reactions by using fluorescence spectroscopy. Treatment of [AuCl(3 b)] with [Ag(ONf)] gave the respective [Au(ONf)(3 b)] only when employing more than 2.5 equivalents of silver salt. The reaction of the “cationic” gold complex with phenyl acetylenes leads to the formation of the respective dinuclear cationic [{(3 b)Au}2(CCPh)]+ and an increase in the level of fluorescence. The rate of the reaction of [Au(ONf)(3 b)] with PhCCH depends on the amount of silver salt in the reaction mixture; a large excess of silver salt accelerates this transformation. In situ fluorescence spectroscopy thus provides valuable information on the association of gold complexes with acetylenes.
A Crabtree-type IrI complex tagged with a fluorescent dye (bodipy) was synthesized. The oxidative addition of H2 converts the weakly fluorescent IrI complex (Φ=0.038) into a highly fluorescent IrIII species (Φ=0.51). This fluorogenic reaction can be utilized for the detection of H2 and to probe the oxidative addition step in the catalytic hydrogenation of olefins.
A Crabtree-type IrI complex tagged with a fluorescent dye (bodipy) was synthesized. The oxidative addition of H2 converts the weakly fluorescent IrI complex (Φ=0.038) into a highly fluorescent IrIII species (Φ=0.51). This fluorogenic reaction can be utilized for the detection of H2 and to probe the oxidative addition step in the catalytic hydrogenation of olefins.
Several metal complexes with a boron dipyrromethene (BODIPY)-functionalized N-heterocyclic carbene (NHC) ligand 4 were synthesized. The fluorescence in [(4)(SIMes)RuCl2(ind)] complex is quenched (Φ=0.003), it is weak in [(4)PdI2(Clpy)] (Φ=0.033), and strong in [(4)AuI] (Φ=0.70). The BODIPY-tagged complexes can experience pronounced changes in the brightness of the fluorophore upon ligand-exchange and ligand-dissociation reactions. Complexes [(4)MX(1,5-cyclooctadiene)] (M=Rh, Ir; X=Cl, I; Φ=0.008–0.016) are converted into strongly fluorescent complexes [(4)MX(CO)2] (Φ=0.53–0.70) upon reaction with carbon monoxide. The unquenching of the Rh and Ir complexes appears to be a consequence of the decreased electron density at Rh or Ir in the carbonyl complexes. In contrast, the substitution of an iodo ligand in [(4)AuI] by an electron-rich thiolate decreases the brightness of the BODIPY fluorophore, rendering the BODIPY as a highly sensitive probe for changes in the coordination sphere of the transition metal.
The present work establishes a new synthetic route that leads to substituted azolium salts. The base stable 1-(4-bromo-2,6-diisopropylphenyl)-3-(2,6-diisopropylphenyl)imidazolidine and 1,3-bis(4-bromo-2,6-diisopropylphenyl)imidazolidine were synthesized and the 4-Br substituents converted into various functional groups through Br/Li exchange or Pd-catalyzed cross-coupling reactions (Suzuki, Sonogashira, and vinylation). The substituted imidazolidines were oxidized, by using chloranil or N-bromosuccinimide, to provide the respective azolium salts, which are convenient precursors to N-heterocyclic carbenes.
1,3,5-Tricycloalkylbenzene (cycloalkyl = C5H9, C6H11) was converted into the respective anilines (by means of nitration and reduction) and then into the corresponding diimines (with glyoxal), the cyclization of which with (HCHO)n/ZnCl2 provided the respective 1,3-bis(2,4,6-tricyclopentylphenyl)imidazolium salt in modest yields. An analogous reaction sequence that employed acenaphthene-1,2-dione instead of glyoxal yielded the two azolium salts in good yields, which were converted into the respective N-heterocyclic carbene (NHC) complexes [(NHC)AgCl], [(NHC)AuCl], [(NHC)RhCl(cod)] (cod = cyclooctadiene), and [(NHC)RhCl(CO)2].
Four 1-(4-R-phenoxy)-2-ethenylbenzenes (R=NMe2, H, Cl, NO2) 4a, 4b, 4c and 4d were reacted with the ruthenium complexes [RuCl2(NHC)(3-phenylindenylidene)(py)] in the presence of a protic resin to result in the formation of the respective Hoveyda-type complexes 5a–d {NHC=SIMes [1,3-bis(2,4,6-trimethylphenylimidazolin)-2-ylidene]} and 6a–d {NHC=SIPr [1,3-bis(2,6-diisopropylphenylimidazolin)-2-ylidene]} in 66–84% yield. The lower steric bulk and the decreased donation of the diaryl ether oxygen atoms in complexes 5 and 6 led to rapidly initiating precatalysts. The Ru(II/III) redox potentials of complexes 6 were determined (6a–d: ΔE=0.89–1.08 V). In the crystal structure of 5b two independent molecules were observed in the unit cell, displaying RuO distances of 226.6(4) and 230.5(3) pm. The catalytic performance of complexes 5 and 6 in various ring-closing metathesis (RCM) reactions was studied. Catalyst loadings of between 15–200 ppm are sufficient for the formation of >90% yield of the respective cyclic products. Complex 6b catalyzes the formation of N-protected 2,5-dihydropyrroles with up to TON 64,000 and TOF 256,000 h−1, of the N-protected 1,2,3,6- tetrahydropyridines with up to TON 18,200 and TOF 73,000 h−1 and of the N-protected 2,3,6,7-tetrahydroazepines with up to TON 8,100 and TOF 32,000 h−1 with yields ranging between 77 and 96%.
Conversion–time data were recorded for various ring-closing metathesis (RCM) reactions that lead to five- or six-membered cyclic olefins by using different precatalysts of the Hoveyda type. Slowly activated precatalysts were found to produce more RCM product than rapidly activated complexes, but this comes at the price of slower product formation. A kinetic model for the analysis of the conversion–time data was derived, which is based on the conversion of the precatalyst (Pcat) into the active species (Acat), with the rate constant kact, followed by two parallel reactions: 1) the catalytic reaction, which utilizes Acat to convert reactants into products, with the rate kcat, and 2) the conversion of Acat into the inactive species (Dcat), with the rate kdec. The calculations employ two experimental parameters: the concentration of the substrate (c(S)) at a given time and the rate of substrate conversion (−dc(S)/dt). This provides a direct measure of the concentration of Acat and enables the calculation of the pseudo-first-order rate constants kact, kcat, and kdec and of kS (for the RCM conversion of the respective substrate by Acat). Most of the RCM reactions studied with different precatalysts are characterized by fast kcat rates and by the kdec value being greater than the kact value, which leads to quasistationarity for Acat. The active species formed during the activation step was shown to be the same, regardless of the nature of different Pcats. The decomposition of Acat occurs along two parallel pathways, a unimolecular (or pseudo-first-order) reaction and a bimolecular reaction involving two ruthenium complexes. Electron-deficient precatalysts display higher rates of catalyst deactivation than their electron-rich relatives. Slowly initiating Pcats act as a reservoir, by generating small stationary concentrations of Acat. Based on this, it can be understood why the use of different precatalysts results in different substrate conversions in olefin metathesis reactions.
Eight new N-Hoveyda-type complexes were synthesized in yields of 67–92 % through reaction of [RuCl2(NHC)(Ind)(py)] (NHC=1,3-bis(2,4,6-trimethylphenylimidazolin)-2-ylidene (SIMes) or 1,3-bis(2,6-diisopropylphenylimidazolin)-2-ylidene (SIPr), Ind=3-phenylindenylid-1-ene, py=pyridine) with various 1- or 1,2-substituted ferrocene compounds with vinyl and amine or imine substituents. The redox potentials of the respective complexes were determined; in all complexes an iron-centered oxidation reaction occurs at potentials close to E=+0.5 V. The crystal structures of the reduced and of the respective oxidized Hoveyda-type complexes were determined and show that the oxidation of the ferrocene unit has little effect on the ruthenium environment. Two of the eight new complexes were found to be switchable catalysts, in that the reduced form is inactive in the ring-opening metathesis polymerization of cis-cyclooctene (COE), whereas the oxidized complexes produce polyCOE. The other complexes are not switchable catalysts and are either inactive or active in both reduced and oxidized states.
Reactions of the Grubbs 3rd generation complexes [RuCl2(NHC)(Ind)(Py)] (N-heterocyclic carbene (NHC)=1,3-bis(2,4,6-trimethylphenylimidazolin)-2-ylidene (SIMes), 1,3-bis(2,6-diisopropylphenylimidazolin)-2-ylidene (SIPr), or 1,3-bis(2,6-diisopropylphenylimidazol)-2-ylidene (IPr); Ind=3-phenylindenylid-1-ene, Py=pyridine) with 2-ethenyl-N-alkylaniline (alkyl=Me, Et) result in the formation of the new N-Grubbs–Hoveyda-type complexes 5 (NHC=SIMes, alkyl=Me), 6 (SIMes, Et), 7 (IPr, Me), 8 (SIPr, Me), and 9 (SIPr, Et) with N-chelating benzylidene ligands in yields of 50–75 %. Compared to their respective, conventional, O-Grubbs–Hoveyda complexes, the new complexes are characterized by fast catalyst activation, which translates into fast and efficient ring-closing metathesis (RCM) reactivity. Catalyst loadings of 15–150 ppm (0.0015–0.015 mol %) are sufficient for the conversion of a wide range of diolefinic substrates into the respective RCM products after 15 min at 50 °C in toluene; compounds 8 and 9 are the most catalytically active complexes. The use of complex 8 in RCM reactions enables the formation of N-protected 2,5-dihydropyrroles with turnover numbers (TONs) of up to 58 000 and turnover frequencies (TOFs) of up to 232 000 h−1; the use of the N-protected 1,2,3,6-tetrahydropyridines proceeds with TONs of up to 37 000 and TOFs of up to 147 000 h−1; and the use of the N-protected 2,3,6,7-tetrahydroazepines proceeds with TONs of up to 19 000 and TOFs of up to 76 000 h−1, with yields for these reactions ranging from 83–92 %.
The reactions of the N,N′-diarylimidazolium and N,N′-diarylimidazolinium salts with chlorosulfonic acid result in the formation of the respective disulfonated N-heterocyclic carbene (NHC) precursors in reasonable yields (46–77%). Water-soluble palladium catalyst complexes, in situ obtained from the respective sulfonated imidazolinium salt, sodium tetrachloropalladate (Na2PdCl4) and potassium hydroxide (KOH) in water, were successfully applied in the copper-free Sonogashira coupling reaction in isopropyl alcohol/water mixtures using 0.2 mol% catalyst loading. The preformed (disulfonatedNHC)PdCl(cinnamyl) complex was used in aqueous Suzuki–Miyaura reactions at 0.1 mol% catalyst loading. The coupling protocol reported here is very useful for Sonogashira reactions of N- and S-heterocyclic aryl bromides and chlorides with aryl- and alkylacetylenes.
Retraction: No Abstract
The palladium complex (0.5 mol-%) of a water-soluble sulfonated fluorenylphosphane (cataCXium Fsulf) enables thefacile Suzuki–Miyaura coupling of various (heterocyclic) aryl tosylates and aryl mesylate with various (heterocyclic) boronic acids in excellent yields (> 95 %) using water as the reaction solvent.
Retraction: The following article from the European Journal of Organic Chemistry, “Suzuki–Miyaura Coupling of Aryl Tosylates and Mesylates in Water”, published online on April 15, 2010 in Wiley Online Library (www.onlinelibrary.wiley.com, doi: 10.1002/ejoc.201000251) and in print (Eur. J. Org. Chem.2010, 2934–2937), has been retracted by agreement between the corresponding author, the journal Editor, Dr. Haymo Ross, and Wiley-VCH. The retraction has been agreed because several 1H and 13C NMR spectroscopic data listed in the manuscript are incorrect, and the original mass spectra cannot be located. Attempts to repeat the synthesis of several representative products under the conditions reported in this manuscript have failed.
Imidazolium salts (NHCewg⋅HCl) with electronically variable substituents in the 4,5-position (H,H or Cl,Cl or H,NO2 or CN,CN) and sterically variable substituents in the 1,3-position (Me,Me or Et,Et or iPr,iPr or Me,iPr) were synthesized and converted into the respective [AgI(NHC)ewg] complexes. The reactions of [(NHC)RuCl2(CHPh)(py)2] with the [AgI(NHCewg)] complexes provide the respective [(NHC)(NHCewg)RuCl2(CHPh)] complexes in excellent yields. The catalytic activity of such complexes in ring-closing metathesis (RCM) reactions leading to tetrasubstituted olefins was studied. To obtain quantitative substrate conversion, catalyst loadings of 0.2–0.5 mol % at 80 °C in toluene are sufficient. The complex with the best catalytic activity in such RCM reactions and the fastest initiation rate has an NHCewg group with 1,3-Me,iPr and 4,5-Cl,Cl substituents and can be synthesized in 95 % isolated yield from the ruthenium precursor. To learn which one of the two NHC ligands acts as the leaving group in olefin metathesis reactions two complexes, [(FL-NHC)(NHCewg)RuCl2(CHPh)] and [(FL-NHCewg)(NHC)RuCl2(CHPh)], with a dansyl fluorophore (FL)-tagged electron-rich NHC ligand (FL-NHC) and an electron-deficient NHC ligand (FL-NHCewg) were prepared. The fluorescence of the dansyl fluorophore is quenched as long as it is in close vicinity to ruthenium, but increases strongly upon dissociation of the respective fluorophore-tagged ligand. In this manner, it was shown for ring-opening metathesis ploymerization (ROMP) reactions at room temperature that the NHCewg ligand normally acts as the leaving group, whereas the other NHC ligand remains ligated to ruthenium.
A method is presented for the high-throughput monitoring of reaction kinetics in homogeneous catalysis, running up to 25 coupling reactions in a single reaction vessel. This method is demonstrated and validated on the Sonogashira reaction, analyzing the kinetics for almost 500 coupling reactions. First, one-pot reactions of phenylacetylene with a set of 20 different meta- and para-substituted aryl bromides were analyzed in the presence of 17 different Pd–phosphine complexes. In addition, the temperature-dependent Sonogashira reactions were examined for 21 different ArX (X=Cl, Br, I) substrates, and the corresponding activation enthalpies and entropies were determined by means of Eyring plots: ArI (ΔH≠=48–62 kJ mol−1; ΔS≠=−71–−39 J mol−1 K; NO2OMe), ArBr (ΔH≠=54–82 kJ mol−1, ΔS≠=−55–11 J mol−1 K), and ArCl (ΔH≠=95–144 kJ mol−1, ΔS≠=−6–100 J mol−1 K). DFT calculations established a linear correlation of ΔH≠ and the Kohn–Sham HOMO energies of ArX (X=Cl, Br, I) and confirmed their involvement in the rate-limiting step. However, despite different CX bond energies, aryl iodides and electron-deficient aryl bromides showed similar activation parameters.
A dicyclohexyl(2-sulfo-9-(3-(4-sulfophenyl)propyl)-9H-fluoren-9-yl)phosphonium salt was synthesized in 64 % overall yield in three steps from simple commercially available starting materials. The highly water-soluble catalyst obtained from the corresponding phosphine and [Na2PdCl4] enabled the Suzuki coupling of a broad variety of N- and S-heterocyclic substrates. Chloropyridines (-quinolines) and aryl chlorides were coupled with aryl-, pyridine- or indoleboronic acids in quantitative yields in water/n-butanol solvent mixtures in the presence of 0.005–0.05 mol % of Pd catalyst at 100 °C, chloropurines were quantitatively Suzuki coupled in the presence of 0.5 mol % of catalyst, and S-heterocyclic aryl chlorides and aryl- or 3-pyridylboronic acids required 0.01–0.05 mol % Pd catalyst for full conversion. The key to the high activity of the Pd-phosphine catalyst is the rational design of the reaction parameters (i.e., the presence of water in the reaction mixture, good solubility of reactants and catalyst in n-butanol/water (3:1), and the electron-rich and sterically demanding nature of the phosphine ligand).
The lithiation/alkylation of fluorene leads to various 9-alkyl-fluorenes (alkyl=Me, Et, iPr, -Pr, -C18H25) in >95 % yields, for which lithiation and reaction with R2PCl (R=Cy, iPr, tBu) generates 9-alkyl, 9-PR2-fluorenes which constitute electron-rich and bulky phosphine ligands. The in-situ-formed palladium–phosphine complexes ([Na2PdCl4], phosphonium salt, base, substrates) were tested in the Sonogashira, Suzuki, and Buchwald–Hartwig reactions of aryl chlorides and aryl bromides in organic solvents. The Sonogashira coupling of aryl chlorides at 100–120 °C leads to >90 % yields with 1 mol % of Pd catalyst. The Suzuki coupling of aryl chlorides typically requires 0.05 mol % of Pd catalyst at 100 °C in dioxane for quantitative product formation. To carry out “green” cross-coupling reactions in water, 9-ethylfluorenyldicyclohexylphosphine was reacted in sulphuric acid to generate the respective 2-sulfonated phosphonium salt. The Suzuki coupling of activated aryl chlorides by using this water-soluble catalyst requires only 0.01 mol % of Pd catalyst, while a wide range of aryl chlorides can be quantitatively converted into the respective coupling products by using 0.1–0.5 mol % of catalyst in pure water at 100 °C. Difficult substrate combinations, such as naphthylboronic acid or 3-pyridylboronic acid and aryl chlorides are coupled at 100 °C by using 0.1–0.5 mol % of catalyst in pure water to obtain the respective N-heterocycles in quantitative yields. The copper-free aqueous Sonogashira coupling of aryl bromides generates the respective tolane derivatives in >95 % yield.
The electron-donating properties of N-heterocyclic carbenes ([N,N′-bis(2,6-dimethylphenyl)imidazol]-2-ylidene and the respective dihydro ligands) with 4,4′-R-substituted aryl rings (4,4′-R=NEt2, OC12H25, Me, H, Br, S(4-tolyl), SO(4-tolyl), SO2(4-tolyl)) were studied. Twelve new N-heterocyclic carbene (NHC) ligands were synthesized as well as the respective iridium complexes [IrCl(cod)(NHC)] and [IrCl(CO)2(NHC)]. Cyclic voltammetry (ΔE1/2) and IR ((CO)) can be used to measure the electron-donating properties of the carbene ligands. Modifying the 4-positions with electron-withdrawing substituents (4-R=−SO2Ar, ΔE1/2=+0.92 V) results in NHC ligands with virtually the same electron-donating capacity as a trialkylphosphine in [IrCl(cod)(PCy3)] (ΔE1/2 =+0.95 V), while [IrCl(cod)(NHC)] complexes with 4-R=NEt2 (ΔE1/2= +0.59 V) show drastically more cathodic redox potentials and significantly enhanced donating properties.
The use of redox-switched phase tags in ferrocenyl-substituted triphenylphosphine combined with DBAD (di-tert-butyl azodicarboxylate) allows high yield (>90 %) Mitsunobu transformations without the need for the chromatographic purification of the products. The redox-switchable phosphine can be easily synthesized in two steps from 4-bromoaniline, ferrocene and chlorodiphenylphosphine. It is separated from the reaction mixture by oxidation with iron(III) chloride and can be recycled efficiently by reductive treatment.
A mixture of Na2PdCl4, CuI and (t-Bu)3PH+BF4− (molar ratio 4 : 3 : 8) dispersed in H2N(i-Pr)2+Br− can be used as a “single source” precatalyst for the Sonogashira coupling of aryl bromides with various aryl- and alkylacetylenes in HN(i-Pr)2 solvent. Arylacetylenes require just 0.005 mol % of Pd catalyst at 80 °C, with TOFs ranging between 3,200 and 10,000 h−1.
Ein Homogenkatalysator wurde mit zwei Ferrocenylgruppen markiert, die sein Löslichkeitsverhalten durch reversibles Schalten zwischen neutralem und dikationischem Zustand bestimmen (siehe Schema). Mithilfe dieses Ansatzes wird die katalytische Aktivität eines Olefinmetathese-Katalysators effektiv an- und ausgeschaltet. Darüber hinaus sind die effiziente Trennung von Katalysator und Produkten sowie eine Wiederverwendung des Katalysators möglich.
Fluorine can do it. Not only oxygen but also fluorine, covalently bonded in organic molecules, is an important donor atom in the coordination chemistry of biorelevant metal ions such as Na+, K+, Mg2+ and Ca2+. Clear evidence in favour of such interactions will be presented in this Minireview, followed by a discussion of possible implications for fluorinated, bioactive compounds.
The Suzuki–Miyaura coupling of aryl chlorides and PhB(OH)2 under biphasic conditions (DMSO/heptane) can be performed in almost quantitative yields over several cycles by means of polymeric Pd catalysts with soluble polyethylene glycol phase tags. Three sterically demanding and electron-rich phosphines 1-CH2Br,4-CH2P(1-Ad)2-C6H4, and 2-PCy2,2′-OH-biphenyl, and 2-PtBu2,2′-OH-biphenyl were covalently bonded to 2000 Dalton MeOPEG-OH. The catalysts, which were formed in situ from Na2[PdCl4], the respective polymeric phosphine, KF/K3PO4, and PhB(OH)2, efficiently couple aryl chlorides at 80 °C at 0.5 mol % loading, resulting in a >90 % yield of the respective biphenyl derivatives. The use of polar phase tags allows the efficient recovery of palladium–phosphine catalysts by simple phase separation of the catalyst-containing DMSO solution and the product-containing n-heptane phase. The high activity (TOF) of the catalyst remains almost constant over more than five reaction cycles, which involve the catalytic reaction, separation of the product phase from the catalyst phase, and addition of new reactants to initiate the next cycle. The Buchwald type biphenyl phosphines form the most active Pd catalysts, which are 1.3–2.8 times more active than catalysts derived from diadamantyl–benzylphosphine, but appear to be less robust in the recycling experiments. There is no apparent leaching of the catalyst into the heptane solution (<0.05 %), as evidenced by spectrophotometric measurements, and contamination of the product with Pd is avoided.
The Sonogashira coupling of various aryl bromides and iodides with different acetylenes was studied under biphasic conditions with soluble, polymer-modified catalysts to allow the efficient recycling of the homogeneous catalyst. For this purpose, several sterically demanding and electron-rich phosphines of the type RPPR2 were synthesised. They are covalently linked to a monomethyl polyethylene glycol ether with a mass of 2000 Dalton (RP=MeOPEG2000) RPPR2: PR2=CH2C6H4CH2P(1-Ad)2, C6H4-P(1-Ad)2, C6H4-PPh2. To couple aryl iodides and acetylenes, the catalyst [(MeCN)2PdCl2]/2 RP-C6H4-PPh2 was used in CH3CN/Et3N/n-heptane (5/2/5). The combined yields of coupling product over five reaction cycles are between 80–95 %. There is no apparent leaching of the catalyst into n-heptane, as evidenced by 1H NMR spectroscopy. The new catalyst [(MeCN)2PdCl2]/2 (1-Ad)2PBn can be used for room-temperature coupling of various aryl bromides and acetylenes in THF with HNiPr2 as a base. A closely related catalyst Na2[PdCl4]/2 RP-CH2C6H4CH2P(1-Ad)2 linked to the polymer was used to couple aryl bromides and acetylenes in DMSO or DMSO/n-heptane at 60 °C with 0.5 mol % Na2[PdCl4], 1 mol % RPPR2 and 0.33 mol % CuI. The combined yield of coupling products over five cycles is always greater than 90 %, except for sterically hindered aryl bromides. The determination of the turnover frequency (TOF) of the catalyst indicates only a small decrease in activity over five cycles. Leaching of the catalyst into the product containing n-heptane solution could not be detected by means of 1H NMR and TXRF; this is indicative of >99.995 % catalyst retention in the DMSO solvent.
Aryl chlorides are suitable substrates for the Sonogashira coupling! By using the versatile catalyst system Na2[PdCl4]/PR3/CuI (PR3=(1-Ad)2PBn, PtBu3), the Sonogashira coupling [Eq. (a)] of aryl chlorides with alkynes generates excellent yields of the corresponding disubstituted aryl alkynes.
The (CH2Br)4 cavitand 1 and its reactions with NaOAc in varying stoichiometric ratios have been used to prepare a series of five different (CH2OAc)4−n(CH2Br)n cavitands (n = 0−3) 2−6 with mixed substituents, which can be separated by column chromatography (combined yield >95%) and are thus available in multi-gram quantities. The substitution reaction is statistical and the yields of individual acetates are controlled by the amount of NaOAc relative to 1. Careful hydrolysis of cavitands 2−6 with LiOH in THF-water results in the formation of the (CH2OH)4−n(CH2Br)n cavitands (n = 0−3) 7−11 in yields of between 70−85%, Treatment of 2−6 with thiourea and NaOH affords the respective (CH2SH)4−n(CH2OH)n cavitands (n = 0−3) 12−16 in yields of around 90%. Reactions of 2−6 with K-Phthalimide generate the respective (CH2Pht.)4−n(CH2OAc)n cavitands (n = 0−3) 17−21, which can be cleaved with hydrazine hydrate resulting in the (CH2NH2)4−n(CH2OH)n cavitands (n = 0−3) 22−26 in an overall yield of 60−70%.
1,1′-(OC2H4OTos)2-ferrocene was treated with various diaza-[n]-crown-m (n/m=12/4, 15/5, 18/6) to give three ferrocene cryptands (n/m=12/4 (FcCrypt), 15/5, 18/6). The complexation of Group 1 and 2 metal ions by FcCrypt leads to large shifts in the redox potentials (up to +500 mV relative to FcCrypt) and consequently to a drastic decrease in the binding strength (up to 108) in the ferrocene cryptands. The redox potential of Fcpda (1,1′-N,N′-bis(dipicol-2-ylamino)-3,3′,4,4′-tetraphenylferrocene) can be modified reversibly by complexation of Zn2+ (E(Fcdpa)=−0.13 V, E(Fcdpa-2 Zn+)=+0.66 V and E(Fcdpa-Zn2+)=+0.72 V). The X-ray crystal structure of FcCrypt-Ca(ClO4)2⋅H2O was determined; Ca2+ is coordinated by six oxygen (Ca2+−O 238.7, 239.1, 239.5, 242.6, 243.6, 247.7 pm) and two nitrogen donors (Ca2+−N 256.1, 259.2 pm) and displays a short Fe−Ca2+ contact (402.7 pm). The stability constants of FcCrypt-Na+ (lg K=8.32 in CH3CN) and FcCrypt-K+ (lg K=4.54 in CH3CN) were determined. The precise adjustment of complex stability and redox potentials of Fcdpa, Fcdpa-Zn2+, FcCrypt (+0.12 V), and FcCrypt-Na+ (+0.395 V) allows coupling of the redox-switchable ferrocene cryptand and the redox-responsive aminoferrocene. In a cyclic process starting from a mixture of Fcdpa+PF6− and FcCrypt-Na+ the addition of Zn(CF3SO3)2 raises the redox potential of Fcdpa+ to that of Fcdpa+-Zn2+. This complex oxidizes FcCrypt-Na+, while the oxidized cryptand displays a drastically reduced affinity towards Na+, so that a mixture containing FcCrypt+, Fcdpa-Zn2+, and uncoordinated Na+ results. The alkali metal ion is recomplexed after cyclam-assisted removal of Zn2+ from the Fcdpa-Zn2+ complex, since Fcdpa is oxidized by FcCrypt+ with reformation of FcCrypt-Na+. Thus two independent chemical processes—the complexation/decomplexation of Zn2+ and of Na+—are linked indirectly with mediation by electron-transfer reactions.
The reaction of 1,1′-ferrocene-bis(methylenepyridinium) salt with 1,4,8,11-tetraazacyclotetradecane-5,12-dione, followed by LiAlH4 reduction results in the formation of FcCyclam. Metal complexes of FcCyclam with M2+=Co2+, Ni2+, Cu2+, and Zn2+ were synthesized from FcCyclam and the respective metal triflates. The complexation of Cu2+ and FcCyclam in CH3CN is preceeded by a rapid electron transfer, followed by a slower complex formation reaction and a reverse electron transfer. The protonation constants of FcCyclam and the stability constants for the Cu2+ complex of FcCyclam (logK=9.26(4) for the formation of the [Cu(FcCyclam)]2+ complex) were determined in 1,4-dioxane/water 70:30 v/v, 0.1 mol dm−3, KNO3, 25 °C. By using FcCyclam one can selectively sense the presence of Cu2+ ions in the presence of Ni2+, Zn2+, Cd2+, Hg2+, and Pb2+ with a very large ΔE≈200 mV, depending on pH. The X-ray crystal structures of FcCyclam and of complexes with Co2+, Ni2+, Cu2+, and Zn2+ were determined and Fe−M2+ distances obtained: Fe−Co2+ 395.9, Fe−Ni2+ 385.4, Fe−Cu2+ 377.7, and Fe−Zn2+ 369.0 pm. The redox potential of FcCyclam is influenced in a characteristic manner by the complexation of M2+. A linear correlation of 1/r≅ΔE [r=distance Fe−M2+ from crystal data, ΔE=E1/2([M(FcCyclam)]2+)−E1/2(FcCyclam)] was found; this is indicative of a mainly Coulomb type interaction between the two metal centers. The nature of the Fe⋅⋅⋅M2+ interaction was also investigated by determining ΔE in several solvents (mixtures) of different dielectric constants ε. The expected relation of ΔE≅1/ε was only found at very high values of ε. At ε<40 increased ion-pairing appears to reduce the effective positive charge at M2+ leading to progessively smaller values of ΔE with lowered ε. The dependence of ΔE and ε can be calculated semiquantitatively by combining the Fuoss ion-pairing theory with the Coulomb model.
The palladium-catalyzed Sonogashira reaction can be used to build optically active, oligomeric 1,2,3-substituted ferrocenes up to the tetramer, as well as polymers, by sequential coupling of optically active (ee>98 %), planar chiral iodoferroceneacetylenes and ferroceneacetylenes. (SFc)-1-Iodoferrocene-2-carbaldehyde (1) was reduced to the alcohol and methylated to give the corresponding methyl ether, which was Sonogashira-coupled with HC≡CSiEt3, resulting in (RFc)-1-(C≡CSiEt3)-2-methoxymethylferrocene (4) (79 %, three steps). Orthometalation with tBuLi followed by quenching with 1,2-diodoethane gave (RFc)-1-(C≡CSiEt3)-2-methoxymethyl-3-iodoferrocene (5). Deprotection of the acetylene with nBu4NF resulted in (RFc)-1-ethynyl-2-methoxymethyl-3-iodoferrocene (6), which was Sonogashira-coupled with itself to produce an optically active polymer. Deprotection of 4 with nBu4NF and Sonogashira coupling of the product with 5 resulted in the dinuclear ferrocene 9. Deprotection of 9 and coupling with 5, followed by deprotection of the resulting acetylene 11, gave the trinuclear ferrocene 12. Another such sequence involving 11 and 5 produced a tetranuclear ferrocene 13. To study the electronic communication in such oligomers in more detail, two symmetrical, closely interrelated, trinuclear ferrocenes 18 and 19 were synthesized. The redox potentials of all the ferrocenes and the ferroceneacetylene polymer were determined by cyclic and square-wave voltammetry. All the metallocenes were investigated by UV/Vis spectroscopy. A linear relationship was found between λmax and 1/n (n=number of ferrocene units in the oligomer). The polymer displayed two redox waves in the cyclic voltammogram, at 0.65 and 0.795 V. The corresponding mixed-valence oligoferrocene cations were synthesized from four ferroceneacetylenes, and their metal-metal charge transfer bands were examined by UV/Vis-NIR. The resonance exchange integrals Had, calculated on the basis of spectral information from the metal-metal charge transfer (MMCT) bands, were between 290 and 552 cm−1.
The fluorine atom of fluorobenzene acts as a normal donor towards hard metal cations (see diagram). This conclusion was drawn on the basis of potentiometric and calorimetric data and X-ray crystal structure determinations. DFT calculations confirm this and predict the Li+–F (fluorobenzene) interaction to be roughly two-thirds as strong as that of Li+ with the oxygen atom of dimethyl ether.