Co-reporter:Yinfeng Yang, Yan Li, Jinghui Wang, Ke Sun, Weiyang Tao, Zhenzhong Wang, Wei Xiao, Yanqiu Pan, Shuwei Zhang, and Yonghua Wang
ACS Chemical Biology May 19, 2017 Volume 12(Issue 5) pp:1363-1363
Publication Date(Web):March 23, 2017
DOI:10.1021/acschembio.6b00762
Globally, cardio-cerebrovascular diseases (CCVDs) are the leading cause of death, and thus the development of novel strategies for preventing and treating such diseases is in urgent need. Traditional Chinese medicine (TCM), used for thousands of years in Asia and other regions, has been proven effective in certain disorders. As a long-time medicinal herb in TCM, Ginkgo biloba leaves (GBLs), have been widely used to treat various diseases including CCVDs. However, the underlying molecular mechanisms of medicinal herbs in treating these diseases are still unclear. Presently, by incorporating pharmacokinetic prescreening, target fishing, and network analysis, an innovative systems-pharmacology platform was introduced to systematically decipher the pharmacological mechanism of action of GBLs for the treatment of CCVDs. The results show that GBLs exhibit a protective effect on CCVDs probably through regulating multiple pathways and hitting on multiple targets involved in several biological pathways. Our work successfully explains the mechanism of efficiency of GBLs for treating CCVDs and, meanwhile, demonstrates that GDJ, an injection generated from GBLs, could be used as a preventive or therapeutic agent in cerebral ischemia. The approach developed in this work offers a new paradigm for systematically understanding the action mechanisms of herb medicine, which will promote the development and application of TCM.
Co-reporter:Jinghui Wang, Yinfeng Yang, Yan Li, and Yonghua Wang
Journal of Agricultural and Food Chemistry 2016 Volume 64(Issue 29) pp:5941-5950
Publication Date(Web):June 29, 2016
DOI:10.1021/acs.jafc.6b01067
Bovine viral diarrhea virus (BVDV) infections are prevailing in cattle populations on a worldwide scale. The BVDV RNA-dependent RNA polymerase (RdRp), as a promising target for new anti-BVDV drug development, has attracted increasing attention. To explore the interaction mechanism of 65 benzimidazole scaffold-based derivatives as BVDV inhibitors, presently, a computational study was performed based on a combination of 3D-QSAR, molecular docking, and molecular dynamics (MD) simulations. The resultant optimum CoMFA and CoMSIA models present proper reliabilities and strong predictive abilities (with Q2 = 0. 64, R2ncv = 0.93, R2pred = 0.80 and Q2 = 0. 65, R2ncv = 0.98, R2pred = 0.86, respectively). In addition, there was good concordance between these models, molecular docking, and MD results. Moreover, the MM-PBSA energy analysis reveals that the major driving force for ligand binding is the polar solvation contribution term. Hopefully, these models and the obtained findings could offer better understanding of the interaction mechanism of BVDV inhibitors as well as benefit the new discovery of more potent BVDV inhibitors.
Co-reporter:Yuan Wang, Zihu Guo, Xuetong Chen, Wenjuan Zhang, Aiping Lu and Yonghua Wang
Molecular BioSystems 2015 vol. 11(Issue 11) pp:3011-3021
Publication Date(Web):12 Aug 2015
DOI:10.1039/C5MB00304K
The determination of cell fate is a key regulatory process for the development of complex organisms that are controlled by distinct genes in mammalian cells. To interpret the decision process in a rigorous, analytical framework, we performed a multi-scale simulation of cell fate decision mediated by the p53 regulatory network in a systems pharmacology framework. The model treats fate determination as a gradual response to stress that delays the initiation of apoptosis to give the cell an opportunity to survive. The newly proposed two-factor model: DNA–p53 coupling explains the phenomenon of the existing biological responses to stress damage for the p53 regulatory network. In addition, the model also reveals that the cell survival rate can be improved by lowering the p53 level in a feedback network to increase its robustness for external stimuli. The present work not only deepens our understanding of cell fate determination, but also provides a theoretical basis for rational drug discovery and development.
Co-reporter:Chunli Zheng;Jinan Wang;Jianling Liu;Mengjie Pei;Chao Huang
Molecular Diversity 2014 Volume 18( Issue 3) pp:621-635
Publication Date(Web):2014 August
DOI:10.1007/s11030-014-9521-y
The term systems pharmacology describes a field of study that uses computational and experimental approaches to broaden the view of drug actions rooted in molecular interactions and advance the process of drug discovery. The aim of this work is to stick out the role that the systems pharmacology plays across the multi-target drug discovery from natural products for cardiovascular diseases (CVDs). Firstly, based on network pharmacology methods, we reconstructed the drug–target and target–target networks to determine the putative protein target set of multi-target drugs for CVDs treatment. Secondly, we reintegrated a compound dataset of natural products and then obtained a multi-target compounds subset by virtual-screening process. Thirdly, a drug-likeness evaluation was applied to find the ADME-favorable compounds in this subset. Finally, we conducted in vitro experiments to evaluate the reliability of the selected chemicals and targets. We found that four of the five randomly selected natural molecules can effectively act on the target set for CVDs, indicating the reasonability of our systems-based method. This strategy may serve as a new model for multi-target drug discovery of complex diseases.
Co-reporter:Xia Wang, Xue Xu, Yan Li, Xiuxiu Li, Weiyang Tao, Bohui Li, Yonghua Wang and Ling Yang
Integrative Biology 2013 vol. 5(Issue 2) pp:351-371
Publication Date(Web):31 Oct 2012
DOI:10.1039/C2IB20204B
Given the imminent threat of influenza pandemics and continuing emergence of new drug-resistant influenza virus strains, novel strategies for preventing and treating influenza disease are urgently needed. Herbal medicine, used for thousands of years in combinational therapies (Herb Formula), plays a significant role in stimulating the host immune system in vivo, and meanwhile, in fighting against the pandemic by directly inhibiting influenza virus in vitro. Such potential Janus functions may spark interest in therapeutic manipulation of virus diseases. Unfortunately, the molecular mechanism of the Janus functions of the medicinal herbs in the treatment of influenza remains unclear. In this work, to illustrate the therapeutic concept of Janus functions in the treatment of influenza, we have introduced a novel systems pharmacology model that integrates pharmacokinetic screening, targeting and network analysis of two representative herbs Lonicera japonica and Fructus Forsythiae that are efficient in the treatment of influenza, inflammation and other diseases. 50 Chemicals with favorable pharmacokinetic profiles have been identified for the two herbs, and the ligand–target network was constructed by complementing the literature-based experimental data deposited in DrugBank. The annotation of these chemicals was assigned using a novel drug targeting approach, and mapped to target–disease and drug–target–pathway networks. The overall data suggest that the medicinal herbs function by indirectly suppressing the virus proliferation via regulating the immune systems in hosts, and also, by directly inhibiting virus proliferation through targeting viral proteins essential for the viral life cycle. For the first time, we have demonstrated the mechanism of medicinal herbs in prevention and treatment of virus diseases via the Janus functions on a systematic level.
Co-reporter:Xue Xu, Xia Wang, Zhentao Xiao, Yan Li and Yonghua Wang
Soft Matter 2012 vol. 8(Issue 2) pp:324-336
Publication Date(Web):25 Oct 2011
DOI:10.1039/C1SM06569F
Protein surface hydration, providing a flexible matrix enabling the protein to respond efficiently to environmental changes, is central to its folding, structure and stability. The aberrant folding of proteins links with a huge variety of diseases, such as prion diseases, diabetes and cancer. Considering the correlation between the mechanism by which a polypeptide chain folds to a specific three-dimensional protein structure, and the role of hydration in the aggregation of misfolded proteins, we use 23 large-scale molecular dynamics simulations to study the local hydration dynamics at the surface of transthyretin (TTR), a thyroid hormone-binding protein that transports thyroxine from the bloodstream to the brain. Monitoring the effects of solvent dynamical behavior around specific mutations by density and spatial distribution entropy maps of the solvent identifies two kinds of water: (1) the hydration water surrounding the stability-bearing mutations characterized by long residential time and slow diffusion; and (2) the water adjacent to the amyloidogenic mutations in fast exchange with the bulk water. Alternative conformations of the protein induced by mutations govern the solvent dynamical behaviors, further evidenced by a map of the spatial distribution entropy of the solvent around the protein. The special behavior of the solvent around these regions is probably crucial in the folding stability and in terms of aggregation loci. These results are also proven by the global perturbations of the protein hydration shell by acidic pH that exhibits dramatic suppression of the aqueous protein motion, and meanwhile, inactivates the stability-bearing waters. This indicates that the acidic medium destroys the cluster structure of water molecules, inducing water diffusion and protein conformational transformation. The present work opens up a possibility of using the mutation and the pH value as probes for protein folding kinetics and functional dynamics measurements, and provides a clue for the treatment of amyloid diseases associated with TTR misfolding.
Co-reporter:Jinan Wang;Fangfang Wang;Zhengtao Xiao;Guowen Sheng
Journal of Molecular Modeling 2012 Volume 18( Issue 7) pp:2943-2958
Publication Date(Web):2012 July
DOI:10.1007/s00894-011-1299-6
The phosphatidylinositol 3-kinase α (PI3Kα) was genetically validated as a promising therapeutic target for developing novel anticancer drugs. In order to explore the structure-activity correlation of benzothiazole series as inhibitors of PI3Kα, comparative molecular field analysis (CoMFA), comparative molecular similarity indices analysis (CoMSIA) were performed on 61 promising molecules to build 3D-QSAR models based on both the ligand- and receptor-based methods. The best CoMFA and CoMSIA models had a cross-validated coefficient rcv2 of 0.618 and 0.621, predicted correlation coefficient rpred2 of 0.812 and 0.83, respectively, proving their high correlative and predictive abilities on both the training and test sets. In addition, docking analysis and molecular dynamics simulation (MD) were also applied to elucidate the probable binding modes of these inhibitors at the ATP binding pocket. Based on the contour maps and MD results, some key structural factors responsible for the activity of this series of compounds were revealed as follows: (1) Ring-A has a strong preference for bulky hydrophobic or aromatic groups; (2) Electron-withdrawing groups at the para position of ring-B and hydrophilic substituents in ring-B region may benefit the potency; (3) A polar substituent like -NHSO2- between ring-A and ring-B can enhance the activity of the drug by providing hydrogen bonding interaction with the protein target. The satisfactory results obtained from this work strongly suggest that the developed 3D-QSAR models and the obtained PI3Kα inhibitor binding structures are reasonable for the prediction of the activity of new inhibitors and be helpful in future PI3Kα inhibitor design.
Co-reporter:Qinfan Li;Xiangya Kong;Zhengtao Xiao;Lihui Zhang
Journal of Molecular Modeling 2012 Volume 18( Issue 6) pp:2279-2289
Publication Date(Web):2012 June
DOI:10.1007/s00894-011-1293-z
Nicotinic acetylcholine receptor (nAChR) is a target for insect-selective neonicotinoid insecticides (NNs), exemplified by imidacloprid (IMI). In the present study, 78 IMI derivatives reported as inhibitors of Drosophila melanogaster nAChR (Dm-nAChR) and Musca domestica nAChR (Md-nAChR) were used for three-dimensional quantitative structure–activity relationship (3D-QSAR) studies. Two optimal models with good predictive power were obtained: Q2 = 0.64, R2pred = 0.72 for Dm-nAChR, and Q2 = 0.63, R2pred = 0.62 for Md-nAChR. In addition, homology modeling, molecular dynamic (MD) simulation, and molecular docking also showed that amino acids located within loops A, C, D and E play key roles in the interaction of Dm-/Md-nAChR with NNs. This is highly consistent with the results of graphical analysis of 3D-QSAR contour plots. Mutation analysis also implicates the Y/S mutation within loop B as being associated closely with NN resistance in Drosophila and Musca. The results obtained lead to a better understanding not only of interactions between these antagonists and Dm-/Md-nAChR, but also of the essential features that should be considered when designing novel inhibitors with desired activities.
Co-reporter:Xue Xu;Jingxuan Fu;Heng Wang;Baidong Zhang;Xia Wang
Ecotoxicology 2011 Volume 20( Issue 2) pp:419-428
Publication Date(Web):2011 March
DOI:10.1007/s10646-011-0593-5
P-glycoprotein (P-gp), as an ATP-binding cassette transporter, transports a wide variety of substrates varying from small molecules like steroids to large polypeptides across the cell membrane in human and animals, even in aquatic animals. Although P-gp protein has attracted much attention of research, its effect on the toxicity of environmental toxicants such as antifouling biocides is still poorly understood. The goal of this study is to evaluate whether copper pyrithione (CuPT), Sea-Nine 211, dichlofluanid and tolylfluanid, four widely used antifouling agents, can be transported by P-gp in embryos of sea urchin Strongylocentrotus intermedius in the presence and absence of the P-gp inhibitor verapamil. Cytotoxcicities of Sea-Nine 211 (EC50 = 99 nM, at 4-arm pluteus) and dichlofluanid (EC50 = 144 nM, at multi-cell) are enhanced by the addition of the P-gp inhibitor, indicating that the two biocides are potential P-gp substrates. Tolylfluanid and CuPT are not transported by P-gp out of the cell, since no obvious changes in the cytotoxicities of the two biocides are observed no matter whether verapamil is added or not. In addition, to understand the mechanisms of ligand binding and its interaction with P-gp, a three-dimensional model of the sea urchin P-gp is generated based on the mouse crystal structure by using homology modeling approach. With this model, a flexible docking is performed and the results indicate that Sea-Nine 211 and dichlofluanid share the same binding site with verapamil, composed of key residues Lys677, Lys753, Thr756, Ala780, Met1033 and Phe1037, whereas tolylfluanid and CuPT display totally different binding modes to P-gp. This further demonstrates that Sea-Nine 211 and dichlofluanid are P-gp substrates, which provides us with new insights into the interactions of P-gp with the antifouling contaminants in aquatic invertebrate embryos.
Co-reporter:Zhengtao Xiao ; Xia Wang ; Xue Xu ; Hong Zhang ; Yan Li
The Journal of Physical Chemistry C 2011 Volume 115(Issue 44) pp:21546-21558
Publication Date(Web):September 16, 2011
DOI:10.1021/jp204017u
Wrapping of single-wall carbon nanotubes (SWCNTs) by single-stranded DNA (ssDNA) was found to be sequence-dependent, offering properties such as the facilitation of SWCN sorting, ultrafast DNA sequencing, and construction of chemical sensors. Although the interactions of nucleic acids with SWCNTs have been studied thoroughly, the DNA–CNT hybrid especially for the oligonucleotides containing more than one nucleotide has not yet been fully understood. To address this, we have examined new and unconventional DNA dinucleotides involving all 16 combinations of two DNA nucleotides attached with chiral (8,4) and armchair (6,6) SWCNTs using all-atom molecular dynamics simulations and thermodynamic analyses. The 16 dinucleotides with different sequence compositions are found to readily adsorb onto SWCNTs and display interesting binding behaviors such as base flipping, local dynamic stability of structure, and conformational shifting. Four dinucleotides, i.e., AC, AG, GC, and GT, share similar dynamic properties (base turning and conformation transformation) in (8,4) and (6,6) systems. The different dynamic profiles between the compositional isomers with the reverse sequences such as the AG and GA show that the sequence order also impacts the dynamic recognition and binding energy of the ssDNA–CNT hybrid. Clustering-analysis-derived representative conformations imply that general dinucleotides are inclined to spread on the SWCNT surface, and the adjacent bases tend to stretch away from each other. Dinucleotides like AC, AT, CG, CT, GC, GG, TA, TC, TG, and TT adopt similar geometries on both CNTs, suggesting that their structures are not predominantly influenced by the nanotube chirality but controlled by the identity of the base sequence, sequence order, and the basic cylindric structure of SWCNT. In addition, the nucleotide bases have a high degree of orientational order on the nanotube surface and the orientations of each base are significantly affected by the sequence of DNA and the chirality of nanotube, emphasizing that the structural order plays an important role in the binding of DNA and CNTs. However, our energy analysis shows that due to small different curvatures of the CNT surface, the binding affinity of most dinucleotides (except AG, CA, CG, and TG) to the chiral and armchair nanotube is not significantly different. Generally, the dinucleotides constituted with purine and thymine exhibit the lowest binding free energy, resulting from the van der Waals interactions and solvent effects. The thymine-based dinucleotides reduce the solvation free energy of the SWCNT in aqueous solution more effectively as compared to other bases. The present work also demonstrates that the total binding free energy is sequence specific but not merely a sum of individual base–SWCNT binding free energies.
Co-reporter:Shao-peng Wei;Zhi-qin Ji;Hui-xiao Zhang;Ji-wen Zhang
Journal of Molecular Modeling 2011 Volume 17( Issue 4) pp:681-693
Publication Date(Web):2011 April
DOI:10.1007/s00894-010-0765-x
For the first time, a set of (43) natural sesquiterpene polyol esters isolated from the root bark of Celastrus angulatus Maxim and Euonymus japonicus Thunb were subjected to 3D-QSAR comparative molecular field analysis (CoMFA) and comparative molecular similarity indices analysis (CoMSIA) studies, with the aim of proposing novel sesquiterpene-based compounds with optimal narcotic or insecticidal activities. The established 3D-QSAR models exhibit reasonable statistical quality and prediction capabilities, with internal cross-validated Q2 values of ∼0.5 and external predicted R2 values of >0.9, respectively. The relative contributions of the steric/electrostatic fields of the 3D-QSAR models show that the electronic effect governs the narcotic activities of the molecules, but the hybrid effect of the electrostatic and hydrophobic interactions is more influential in the insecticidal activities of the compounds. These findings may have valuable implications for the development of novel natural insecticides.
Co-reporter:Lihui Zhang, Tianjun Liu, Xia Wang, Jinan Wang, Guohui Li, Yan Li, Ling Yang, Yonghua Wang
Biosystems (January 2014) Volume 115() pp:
Publication Date(Web):1 January 2014
DOI:10.1016/j.biosystems.2013.04.003
The interaction of 278 monocyclic and bicyclic pyrimidine derivatives with human A2A adenosine receptor (AR) was investigated by employing molecular dynamics, thermodynamic analysis and three-dimensional quantitative structure–activity relationship (3D-QSAR) approaches. The binding analysis reveals that the pyrimidine derivatives are anchored in TM2, 3, 5, 6 and 7 of A2A AR by the aromatic stacking and hydrogen bonding interactions. The key residues involving Phe168, Glu169, and Asn253 stabilize the monocyclic and bicyclic cores of inhibitors. The thermodynamic analysis by molecular mechanics/Poisson Boltzmann surface area (MM-PBSA) approach also confirms the reasonableness of the binding modes. In addition, the ligand-/receptor-based comparative molecular similarity indices analysis (CoMSIA) models of high statistical significance were generated and the resulting contour maps correlate well with the structural features of the antagonists essential for high A2A AR affinity. A minor/bulky group with negative charge at C2/C6 of pyrimidine ring respectively enhances the activity for all these pyrimidine derivatives. Particularly, the higher electron density of the ring in the bicyclic derivatives, the more potent the antagonists. The obatined results might be helpful in rational design of novel candidate of A2A adenosine receptor antagonist for treatment of Parkinson's disease.
Co-reporter:X. Xu, Z. Ma, X. Wang, Z.T. Xiao, Y. Li, Z.H. Xue, Y.H. Wang
Journal of Structural Biology (February 2012) Volume 177(Issue 2) pp:358-366
Publication Date(Web):1 February 2012
DOI:10.1016/j.jsb.2011.12.008
Soluble proteins with amyloidogenic propensity such as the tumor suppressor protein p53 have high proportion of incompletely desolvated backbone H bonds (HB). Such bonds are vulnerable to water attack, thus potentially leading to the misfolding of these proteins. However, it is still not clear how the surrounding solvent influences the protein native states. To address this, systematic surveys by molecular dynamics simulations and entropy analysis were performed on the p53 core domain in this work. We examined seven wild/mutant X-ray structures and observed two types of water-network hydration in three “hot hydration centers” (DNA- or small molecule- binding surfaces of the p53 core domain). The “tight” water, resulting from the local collective hydrogen-bond interactions, is probably fundamental to the protein structural stability. The second type of water is highly “dynamical” and exchanges very fast within the bulk solution, which is unambiguously assisted by the local protein motions. An entropy mapping of the solvent around the protein and a temperature perturbation analysis further present the main features of the p53 hydration network. The particular environment created by different water molecules around the p53 core domain also partly explains the structural vulnerabilities of this protein.
Co-reporter:Hong Zhang, Yao Yao, Huibin Yang, Xia Wang, Zhuo Kang, Yan Li, Guohui Li, Yonghua Wang
Insect Biochemistry and Molecular Biology (August 2012) Volume 42(Issue 8) pp:583-595
Publication Date(Web):1 August 2012
DOI:10.1016/j.ibmb.2012.04.005
As one potent plant protease inhibitor, potato carboxypeptidase inhibitor (PCI) can competitively inhibit insect digestive metallocarboxypeptidases (MCPs) through interfering with its digestive system that causes amino acid deficiencies and leading to serious developmental delay and mortality. However, this effective biological pest control is significantly impaired by the PCI-resistant insect MCPs. Therefore, deep understanding of the resistant mechanism of insect MCPs is particularly necessary for designing new durable pest control regimen and developing effective pesticides. In this study, the binding of PCI and small molecular inhibitor THI to insect PCI-sensitive/-resistant MCPs and human MCP was investigated by docking, molecular dynamics (MD) simulations and thermodynamic analysis. The structural analysis from MD simulations indicates that the PCI-resistant mechanism of CPBHz is mainly dominated by the Trp277A, which changes the conformation of β8-α9 loop and therefore narrow the access to the active site of CPBHz, prohibiting the entrance of the C termini tail of PCI. Additionally, the insertion of Gly247A weakens the stabilization of CPBHz and PCI through disrupting the hydrogen bond formation with its surrounding residues. Furthermore, the predicted binding free energies gives explanation of structure affinity relationship of PCI and THI with MCPs and suggest that the electrostatic energy is the main contribution term affecting the difference in binding affinities. Finally, the decomposition analysis of binding free energies infers that the key residues Glu72, Arg127, Ile247/Leu247 and Glu270 are critical for the binding of PCI/THI to MCPs.Graphical abstractDownload high-res image (312KB)Download full-size imageHighlights► The Y277AW mutation dominates the PCI-resistant mechanism of CPBHz. ► Binding free energies discriminate the PCI-sensitive/-insensitive insect MCPs. ► Small molecular inhibitor-MCPs study provides useful information.
Co-reporter:Xia Wang, Yan Li, Xue Xu, Yong-hua Wang
Biosystems (April 2010) Volume 100(Issue 1) pp:31-38
Publication Date(Web):April 2010
DOI:10.1016/j.biosystems.2009.12.005
Co-reporter:Wei Zhou, Chao Huang, Yan Li, Jinyou Duan, Yonghua Wang, Ling Yang
Toxicology (8 February 2013) Volume 304() pp:173-184
Publication Date(Web):8 February 2013
DOI:10.1016/j.tox.2012.12.012
Although the assessment of toxicity of various agents, -omics (genomic, proteomic, metabolomic, etc.) data has been accumulated largely, the acquirement of toxicity information of variety of molecules through experimental methods still remains a difficult task. Presently, a systems toxicology approach that integrates massive diverse chemical, genomic and toxicological information was developed for prediction of the toxin targets and their related networks. The procedures are: (1) by use of two powerful statistical methods, i.e., support vector machine (SVM) and random forest (RF), a systemic model for prediction of multiple toxin–target interactions using the extracted chemical and genomic features has been developed with its reliability and robustness estimated. And the qualitative classification of targets according to the phenotypic diseases has been taken into account to further uncover the biological meaning of the targets, as well as to validate the robustness of the in silico models. (2) Based on the predicted toxin–target interactions, a genome-scale toxin–target-disease network exampled by cardiovascular disease is generated. (3) A topological analysis of the network is carried out to identify those targets that are most susceptible in human to topical agents including the most critical toxins, as well as to uncover both the toxin-specific mechanisms and pathways. The methodologies presented herein for systems toxicology will make drug development, toxin environmental risk assessment more efficient, acceptable and cost-effective.
Co-reporter:Tianli Pei, Chunli Zheng, Chao Huang, Xuetong Chen, Zihu Guo, Yingxue Fu, Jianling Liu, Yonghua Wang
Journal of Ethnopharmacology (22 August 2016) Volume 190() pp:272-287
Publication Date(Web):22 August 2016
DOI:10.1016/j.jep.2016.06.001
Ethnopharmacological relevanceVitiligo is a depigmentation disorder, which results in substantial cosmetic disfigurement and poses a detriment to patients' physical as well as mental. Now the molecular pathogenesis of vitiligo still remains unclear, which leads to a daunting challenge for vitiligo therapy in modern medicine. Herbal medicines, characterized by multi-compound and multi-target, have long been shown effective in treating vitiligo, but their molecular mechanisms of action also remain ambiguous.Materials and methodsHere we proposed a systems pharmacology approach using a clinically effective herb formula as a tool to detect the molecular pathogenesis of vitiligo. This study provided an integrative analysis of active chemicals, drug targets and interacting pathways of the Uygur medicine Qubaibabuqi formula for curing Vitiligo.ResultsThe results show that 56 active ingredients of Qubaibabuqi interacting with 83 therapeutic proteins were identified. And Qubaibabuqi probably participate in immunomodulation, neuromodulation and keratinocytes apoptosis inhibition in treatment of vitiligo by a synergistic/cooperative way.ConclusionsThe drug-target network-based analysis and pathway-based analysis can provide a new approach for understanding the pathogenesis of vitiligo and uncovering the molecular mechanisms of Qubaibabuqi, which will also facilitate the application of traditional Chinese herbs in modern medicine.Download high-res image (139KB)Download full-size image
Co-reporter:Wei Zhou, Chao Huang, Yan Li, Jinyou Duan, Yonghua Wang, Ling Yang
Toxicology (20 March 2014) Volume 317() pp:58
Publication Date(Web):20 March 2014
DOI:10.1016/j.tox.2014.01.001
Co-reporter:Wei Zhou, Yonghua Wang
Journal of Ethnopharmacology (10 January 2014) Volume 151(Issue 1) pp:66-77
Publication Date(Web):10 January 2014
DOI:10.1016/j.jep.2013.11.007
Ethnopharmacological relevanceCoronary heart disease (CAD) is one of the most dangerous threats to human health due to its high incidence and high mortality. CAD has several major types, such as blood stasis and qi deficiency according to the syndromes of diagnosis in traditional Chinese medicine (TCM), which are treated with different herbs or compound prescriptions. However, up to now a deep analysis of the relationship between CAD and its types both at molecular or systems levels is still unavailable, which greatly limits the combination of TCMs with Western drugs to form an integrative/alternative medicine for treatment of the complex disease.Materials and methodsIn this review, we attempt to decipher the underlying mechanisms of major types of CAD by connecting the drugs, targets and diseases to obtain the compound-target-disease associations for reconstructing the biologically-meaningful networks based on systems pharmacology method.ResultsThe results indicate that the herbs for eliminating blood stasis have pharmacological activity of dilating blood vessel, improving the microcirculation, reducing blood viscosity and regulating blood lipid, while qi-enhancing herbs have the potential for enhancing energy metabolism and anti-inflammation.ConclusionsA systematic exploration of types of CAD may bring out the best between research on drug molecules and TCM phenotypic information, so as to accelerate development of network-based drug discovery as well as to facilitate the therapy of this disease.Coronary heart disease (CAD) is one of the most dangerous threats to human health due to its high incidence and high mortality. CAD has several major types, such as blood stasis and qi deficiency, according to the syndromes of diagnosis in traditional Chinese medicine (TCM), which are treated with different herbs or compound prescriptions. The current review aims to decipher the underlying connections between the major types and CAD in the TCM framework. We attempt to take the known herbs as the probe to uncover the potential mechanism between the major types and CAD by connecting the drugs, targets and diseases so as to obtain compound-target-disease associations for reconstructing the biologically-meaningful networks. For example, we take the three key herbs for blood stasis treatment as an example to explore the pharmacological mechanism of treatment of blood stasis of CAD.Download high-res image (289KB)Download full-size image
Co-reporter:Peng Li, Jianxin Chen, Jinan Wang, Wei Zhou, Xia Wang, Bohui Li, Weiyang Tao, Wei Wang, Yonghua Wang, Ling Yang
Journal of Ethnopharmacology (10 January 2014) Volume 151(Issue 1) pp:93-107
Publication Date(Web):10 January 2014
DOI:10.1016/j.jep.2013.07.001
Ethnopharmacological relevanceMulti-target therapeutics is a promising paradigm for drug discovery which is expected to produce greater levels of efficacy with fewer adverse effects and toxicity than monotherapies. Medical herbs featuring multi-components and multi-targets may serve as valuable resources for network-based multi-target drug discovery.Materials and methodsIn this study, we report an integrated systems pharmacology platform for drug discovery and combination, with a typical example applied to herbal medicines in the treatment of cardiovascular diseases.ResultsFirst, a disease-specific drug–target network was constructed and examined at systems level to capture the key disease-relevant biology for discovery of multi-targeted agents. Second, considering an integration of disease complexity and multilevel connectivity, a comprehensive database of literature-reported associations, chemicals and pharmacology for herbal medicines was designed. Third, a large-scale systematic analysis combining pharmacokinetics, chemogenomics, pharmacology and systems biology data through computational methods was performed and validated experimentally, which results in a superior output of information for systematic drug design strategies for complex diseases.ConclusionsThis strategy integrating different types of technologies is expected to help create new opportunities for drug discovery and combination.Download high-res image (123KB)Download full-size image
Co-reporter:Yingxue Fu, Yonghua Wang, Boli Zhang
Journal of Traditional Chinese Medical Sciences (1 October 2014) Volume 1(Issue 2) pp:84-91
Publication Date(Web):1 October 2014
DOI:10.1016/j.jtcms.2014.09.006
Identified as a treasure of natural herbal products, traditional Chinese medicine (TCM) has attracted extensive attention for their moderate treatment effect and lower side effect. Cardio-cerebrovascular diseases (CCVD) are a leading cause of death. TCM is used in China to prevent and treat CCVD. However, the complexity of TCM poses challenges in understanding the mechanisms of herbs at a systems-level, thus hampering the modernization and globalization of TCM. A novel model, termed traditional Chinese medicine systems pharmacology (TCMSP) analysis platform, which relies on the theory of systems pharmacology and integrates absorption, distribution, metabolism, excretion and toxicity (ADME/T) evaluation, target prediction and network/pathway analysis, was proposed to address these problems. Here, we review the development of systems pharmacology, the TCMSP approach and its applications in the investigations of CCVD and compare it with other methods. TCMSP assists in uncovering the mechanisms of action of herbal formulas used in treating CCVD. It can also be applied in ascertaining the different syndrome patterns of coronary artery disease, decoding the multi-scale mechanisms of herbs, and in understanding the mechanisms of herbal synergism.



![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2.png)
![5-Heptenoic acid,7-[(2R,3S,4S)-tetrahydro-4,6-dihydroxy-2-[(1E,3S)-3-hydroxy-1-octen-1-yl]-2H-pyran-3-yl]-,(5Z)-](http://img.cochemist.com/ccimg/54400/54397-85-2_b.png)
![N-[4-(5-NITRO-1H-BENZIMIDAZOL-2-YL)PHENYL]ACETAMIDE](http://img.cochemist.com/ccimg/745900/745809-88-5.png)
![N-[4-(5-NITRO-1H-BENZIMIDAZOL-2-YL)PHENYL]ACETAMIDE](http://img.cochemist.com/ccimg/745900/745809-88-5_b.png)
![1-Piperidineacetamide, N-[4-(1H-benzimidazol-2-yl)phenyl]-](http://img.cochemist.com/ccimg/210300/210221-97-9.png)
![1-Piperidineacetamide, N-[4-(1H-benzimidazol-2-yl)phenyl]-](http://img.cochemist.com/ccimg/210300/210221-97-9_b.png)
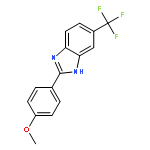
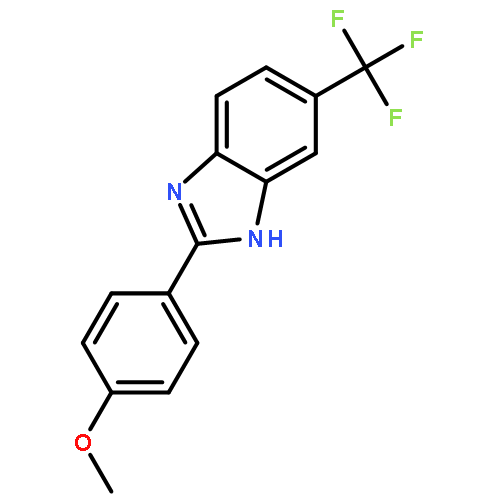
![4-(5-Nitro-1H-benzo[d]imidazol-2-yl)aniline](http://img.cochemist.com/ccimg/71100/71002-88-5.png)
![4-(5-Nitro-1H-benzo[d]imidazol-2-yl)aniline](http://img.cochemist.com/ccimg/71100/71002-88-5_b.png)


![Acetamide, N-[4-(1H-benzimidazol-2-yl)phenyl]-](http://img.cochemist.com/ccimg/27100/27030-98-4.png)
![Acetamide, N-[4-(1H-benzimidazol-2-yl)phenyl]-](http://img.cochemist.com/ccimg/27100/27030-98-4_b.png)


![4-(1H-Benzo[d]imidazol-2-yl)aniline](http://img.cochemist.com/ccimg/3000/2963-77-1.png)
![4-(1H-Benzo[d]imidazol-2-yl)aniline](http://img.cochemist.com/ccimg/3000/2963-77-1_b.png)