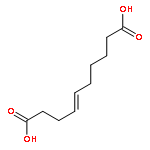
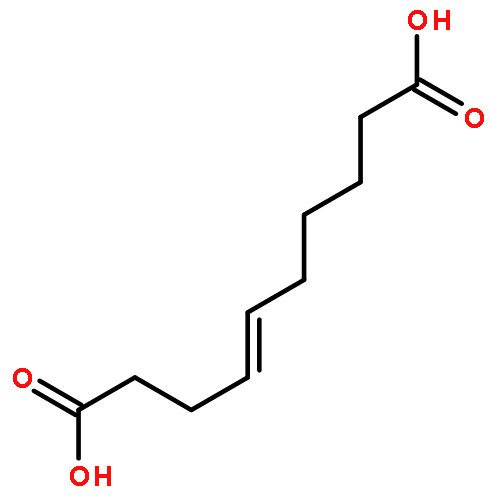
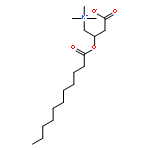
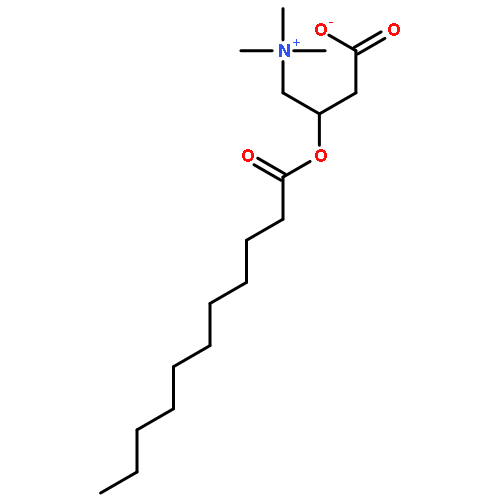
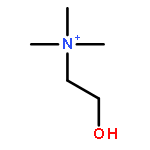
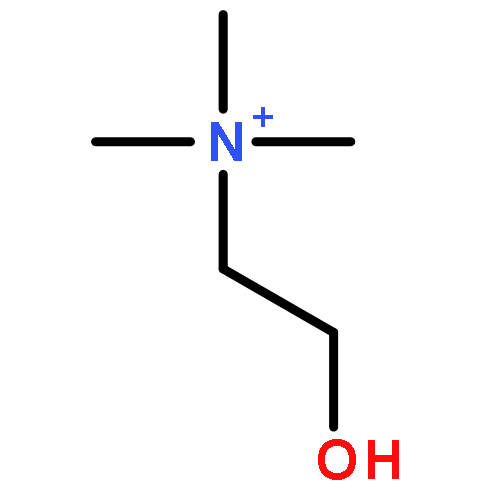
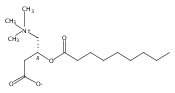
Co-reporter:Zhonghua Wang;Yajie Zheng;Yanping Zhang;Baoxin Zhao;Zhe Liu;Jing Xu;Yanhua Chen;Zhao Yang;Fenfen Wang;Huiqing Wang;Ruiping Zhang;Jiuming He
Journal of Proteome Research June 5, 2015 Volume 14(Issue 6) pp:2583-2593
Publication Date(Web):2017-2-22
DOI:10.1021/acs.jproteome.5b00134
The toxicities of polycyclic aromatic hydrocarbons (PAHs) have been extensively explored due to their carcinogenic and mutagenic potency; however, little is known about the metabolic responses to chronic environmental PAH exposure among the general population. In the present study, 566 healthy volunteers were dichotomized into exposed and control groups to investigate PAH-induced perturbations in the metabolic profiles. Nine urine PAH metabolites were measured by a sensitive LC–MS/MS method to comprehensively evaluate the PAH exposure level of each individual, and the metabolic profiles were characterized via a LC–MS-based metabolomic approach. PAH exposure was correlated to its metabolic outcomes by linear and logistic regression analyses. Metabolites related to amino acid, purine, lipid, and glucuronic acid metabolism were significantly changed in the exposed group. 1-Hydroxyphenanthrene and dodecadienylcarnitine have potential as sensitive and reliable biomarkers for PAH exposure and its metabolic outcomes, respectively, in the general population. These findings generally support the hypothesis that environmental PAH exposure causes oxidative stress-related effects in humans. The current study provides new insight into the early molecular events induced by PAH exposure in the actual environment.Keywords: environmental health; Metabolomics; molecular epidemiology; oxidative stress; PAH exposure;
Co-reporter:Zhuoling An;Ruiping Zhang;Yanhua Chen;Yongmei Song;Qimin Zhan;Jianghao Sun;Lijia Dong;Jiuming He;Jinfa Bai
Journal of Proteome Research August 6, 2010 Volume 9(Issue 8) pp:4071-4081
Publication Date(Web):2017-2-22
DOI:10.1021/pr100265g
An integrated ionization approach of electrospray ionization (ESI), atmospheric pressure chemical ionization (APCI), and atmospheric pressure photoionization (APPI) combining with rapid resolution liquid chromatography mass spectrometry (RRLC−MS) has been developed for performing global metabonomic analysis on complex biological samples. This approach was designed to overcome the low ionization efficiencies of endogenous metabolites due to diverse physicochemical properties as well as ion suppression, and obtain comprehensive metabolite profiles in LC−MS analysis. Ionization capability and applicability were manifested by improved ionization efficiency and enlarged metabolite coverage in analysis on typical urinary metabolite standards and urine samples from healthy volunteers. The method was validated by the limit of detection and precision. When applied to the global metabonomic studies of lung cancer, more comprehensive biomarker candidates were obtained to reflect metabolic traits between healthy volunteers and lung cancer patients, including 74 potential biomarkers in positive ion mode and 59 in negative ion mode. Taking identical potential biomarkers of any two or three ionization methods into account, analysis using ESI-MS in positive (+) and negative (−) ion mode contributed to 70 and 64% of the total potential biomarkers, respectively. The biomarker discovery capability of (±) APCI-MS accounted for 45 and 42% of the overall; meanwhile (±) APPI-MS amounted for 39 and 54%. These results indicated that potential biomarkers with vital biological information could be missed if only a single ionization method was used. Furthermore, 11 potential biomarkers were identified including amino acids, nucleosides, and a metabolite of indole. They revealed elevated amino acid and nucleoside metabolism as well as protein degradation in lung cancer patients. This proposed approach provided a more comprehensive picture of the metabolic changes and further verified identical biomarkers that were obtained simultaneously using different ionization methods.Keywords: APPI; ESI, APCI; integrated ionization approach; lung cancer; metabonomics; RRLC−MS, potential biomarkers;
Co-reporter:Guoqing Shen;Jianghao Sun;Yanhua Chen;Ruiping Zhang;Yaping Tian;Yongmei Song;Jiuming He;Yi Zhang;Xiaoguang Chen
Journal of Proteome Research April 1, 2011 Volume 10(Issue 4) pp:1953-1961
Publication Date(Web):Publication Date (Web): January 28, 2011
DOI:10.1021/pr101198q
A metabonomic approach based on complementary hydrophilic interaction chromatography and reversed-phase liquid chromatography combined with tandem mass spectrometry and time-course analysis of metabolites was implemented to find more reliable potential biomarkers in urine of Walker 256 tumor-bearing rats. A major challenge in metabonomics is distinguishing reliable biomarkers that are closely associated with the genesis and progression of diseases from those that are unrelated but altered significantly. In this study, these biomarkers were selected according to the change trends of discriminating metabolites during the genesis and progression of cancer. Seven consecutive batches of urine samples from preinoculation to 16 days after were collected and analyzed. Multivariate analysis revealed 87 discriminating metabolites. Time-course analysis of discriminating metabolites was used to select more reliable biomarkers with regular and reasonable change trends. Finally, 47 were found and 15 were identified including 12 carnitine derivatives, 2 amino acid derivatives, 1 nucleoside. On the basis of time-course behaviors of these potential biomarkers, we hypothesize such disruption might result from elevated cell proliferation, reduced β-oxidation of fatty acids, and poor renal tubular reabsorption. These studies demonstrate that this method can help to find more reliable potential biomarkers and provide valuable biochemical insights into metabolic alterations in tumor-bearing biosystems.Keywords (keywords): biomarkers; hydrophilic interaction chromatography; metabonomics; reversed-phase liquid chromatography; time-course analysis; Walker 256 tumor;
Co-reporter:Huiqing Wang, Jing Xu, Yanhua Chen, Ruiping Zhang, Jiuming He, Zhonghua Wang, Qingce Zang, Jinfeng Wei, Xiaowei Song, and Zeper Abliz
Analytical Chemistry 2016 Volume 88(Issue 7) pp:3459
Publication Date(Web):March 7, 2016
DOI:10.1021/acs.analchem.5b04709
Sample preparation is a critical step in tissue metabolomics. Therefore, a comprehensive and systematic strategy for the screening of tissue preparation protocols is highly desirable. In this study, we developed an Optimization and Evaluation Strategy based on LC–MS to screen for a high-extractive efficiency and reproducible esophageal tissue preparation protocol for different types of endogenous metabolites (amino acids, carnitines, cholines, etc.), with a special focus on low-level metabolites. In this strategy, we first selected a large number of target metabolites based on literature survey, previous work in our lab, and known metabolic pathways. For these target metabolites, we tested different solvent extraction methods (biphasic solvent extraction, two-step [TS], stepwise [SW], all-in one [AO]; single-phase solvent extraction, SP) and esophageal tissue disruption methods (homogenized wet tissue [HW], ground wet tissue [GW], and ground dry tissue [GD]). A protocol involving stepwise addition of solvents and a homogenized wet tissue protocol (SWHW) was superior to the others. Finally, we evaluated the stability of endogenous metabolites in esophageal tissues and the sensitivity, reproducibility, and recovery of the optimal protocol. The results proved that the SWHW protocol was robust and adequate for bioanalysis. This strategy will provide important guidance for the standardized and scientific investigation of tissue metabolomics.
Co-reporter:Yan Gao, Ruiping Zhang, Jinfa Bai, Xuejun Xia, Yanhua Chen, Zhigang Luo, Jing Xu, Yang Gao, Yuling Liu, Jiuming He, and Zeper Abliz
Analytical Chemistry 2015 Volume 87(Issue 15) pp:7535
Publication Date(Web):July 1, 2015
DOI:10.1021/acs.analchem.5b01205
Detection and identification of unknown or low-level drug-related metabolites in complex biological materials is an ongoing challenge. A highly selective and sensitive method could be a possible solution. Here, we proposed a targeted data-independent acquisition and mining (TDIAM) strategy for the rapid identification of trace drug metabolites using ultra-high-performance liquid chromatography coupled with high-resolution tandem mass spectrometry (UHPLC-HRMS/MS). In this strategy, raw data is acquired by a novel tm-MS scan, which contains an interleaved full MS scan with a targeted mass range and a product ion scan by selecting all ions in the targeted mass range as precursor ions. For efficient discovery of metabolites, raw data are analyzed by a new postacquisition processing method, Molecule- and Fragmentation-driven Mass Defect Filters (MF-MDFs), which was developed based on the fragmentation of parent drug to pick out molecular ions and fragment ions of potential metabolites from the complex matrix. When applying the proposed strategy to paclitaxel metabolism research, we successfully identified 10 metabolites, among which six were not previously reported. The results demonstrated that TDIAM greatly improved throughput, detective sensitivity, and selectivity and, more importantly, yielded almost the same spectrum as traditional HRMS/MS. Therefore, TDIAM provides structure-enriched evidence to confirm the existence and elucidate the structures of metabolites. This strategy is suitable for identification of metabolites present at low concentrations in a complex matrix, and it has the potential to provide an efficient, sensitive, and labor-saving solution for drug metabolite research.
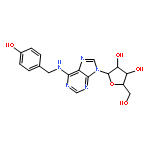
Co-reporter:Jingjing He, Zhigang Luo, Lan Huang, Jiuming He, Yi Chen, Xianfang Rong, Shaobo Jia, Fei Tang, Xiaohao Wang, Ruiping Zhang, Jianjun Zhang, Jiangong Shi, and Zeper Abliz
Analytical Chemistry 2015 Volume 87(Issue 10) pp:5372
Publication Date(Web):April 15, 2015
DOI:10.1021/acs.analchem.5b00680
Elucidation of the mechanism of action for drug candidates is fundamental to drug development, and it is strongly facilitated by metabolomics. Herein, we developed an imaging metabolomics method based on air-flow-assisted desorption electrospray ionization mass spectrometry imaging (AFADESI-MSI) under ambient conditions. This method was subsequently applied to simultaneously profile a novel anti-insomnia drug candidate, N6-(4-hydroxybenzyl)-adenosine (NHBA), and various endogenous metabolites in rat whole-body tissue sections after the administration of NHBA. The principal component analysis (PCA) represented by an intuitive color-coding scheme based on hyperspectral imaging revealed in situ molecular profiling alterations in response to stimulation of NHBA, which are in a very low intensity and hidden in massive interferential peaks. We found that the abundance of six endogenous metabolites changed after drug administration. The spatiotemporal distribution indicated that five altered molecules—including neurotransmitter γ-aminobutyric acid, neurotransmitter precursors choline and glycerophosphocholine, energy metabolism-related molecules adenosine (an endogenous sleep factor), and creatine—are closely associated with insomnia or other neurological disorders. These findings not only provide insights into a deep understanding on the mechanism of action of NHBA, but also demonstrate that the AFADESI-MSI-based imaging metabolomics is a powerful technique to investigate the molecular mechanism of drug action, especially for drug candidates with multitarget or undefined target in the preclinical study stage.
Co-reporter:Qingce Zang, Yang Liu, Jiuming He, Xiaofei Yue, Ruiping Zhang, Renzhi Wang, Zeper Abliz
Journal of Chromatography B 2015 Volume 989() pp:91-97
Publication Date(Web):1 May 2015
DOI:10.1016/j.jchromb.2015.03.004
•In this study, a sensitive, rapid and high-throughput quantitative bioanalytical method has been established by using HPLC–MS/MS for the determination of bromocriptine at trace level in human prolactinoma tissue. As little as 20 mg (wet weight) tissue sample was required and total analysis time was 6 min in this method. We utilized this HPLC–MS/MS method to analyze the distribution difference of bromocriptine in post-operative responsive and non-responding prolactinomas. Finally we have found that bromocriptine concentration in non-responding prolactinomas was significantly higher than that in sensitive prolactinomas, and confirmed that the failure of bromocriptine treatment in non-responding patients with prolactinoma was “intrinsic” tumor (cell) resistence, rather than insufficient drug concentration in tumor tissue.Usually, insufficient intratumoral concentration of therapeutic drugs is one of the reasons for tumor treatment failure. However, little is known about intratumoral distribution of bromocriptine in non-responding prolactinomas because of extremely low drug concentration and small prolactinoma tissue samples. In this study, a sensitive, rapid and high-throughput quantitative bioanalytical method has been established by using high-performance liquid chromatography–tandem mass spectrometry (HPLC–MS/MS) for the determination of bromocriptine at trace level in human prolactinoma tissue. As little as 20 mg (wet weight) tissue sample was required and total analysis time was 6 min in this method. The assay quantifies over a linear range of 50 fg/mg to 5 pg/mg, and has a 25 fg/mg limit of detection at a signal/noise ratio of 3. This validated method was successfully used to quantitatively determine bromocriptine in clinical post-operative bromocriptine-sensitive and -resistant prolactinomas. The results revealed bromocriptine concentration in resistant prolactinomas (0.49–1.25 pg/mg) was significantly higher than that in sensitive prolactinomas (0.057–0.47 pg/mg). These results provided direct evidence to demonstrate the reseaon for failure of bromocriptine treatment in some patients with prolactinoma was “intrinsic” tumor (cell) resistence, rather than insufficient drug concentration in tumor tissue. Additionaly, this HPLC–MS/MS method has been shown to be suitable for bromocriptine analysis in small amount tissue sample and could be adapted for therapeutic drug monitoring of other clinical medicine.
Co-reporter:Yanhua Chen, Guoqing Shen, Ruiping Zhang, Jiuming He, Yi Zhang, Jing Xu, Wei Yang, Xiaoguang Chen, Yongmei Song, and Zeper Abliz
Analytical Chemistry 2013 Volume 85(Issue 16) pp:7659
Publication Date(Web):July 15, 2013
DOI:10.1021/ac401400b
It is essential to choose one preprocessing method for liquid chromatography–mass spectrometry (LC-MS)-based metabolomics studies of urine samples in order to overcome their variability. However, the commonly used normalization methods do not substantially reduce the high variabilities arising from differences in urine concentration, especially for signal saturation (abundant metabolites exceed the dynamic range of the instrumentation) or missing values. Herein, a simple preacquisition strategy based on differential injection volumes calibrated by creatinine (to reduce the concentration differences between the samples), combined with normalization to “total useful MS signals” or “all MS signals”, is proposed to overcome urine variabilities. This strategy was first systematically compared with other popular normalization methods by application to serially diluted urine samples. Then, the method has been verified using rat urine samples of pre- and postinoculation of Walker 256 carcinoma cells. The results showed that the calibration of injection volumes based on creatinine values could effectively eliminate intragroup differences caused by variations in the concentrations of urinary metabolites, thus giving better parallelism and clustering effects. In addition, peak area normalization could further eliminate intraclass differences. Therefore, the strategy of combining peak area normalization with calibration of injection volumes of urine samples based on their creatinine values is effective for solving problems associated with urinary metabolomics.
Co-reporter:Zhigang Luo, Jiuming He, Yi Chen, Jingjing He, Tao Gong, Fei Tang, Xiaohao Wang, Ruiping Zhang, Lan Huang, Lianfeng Zhang, Haining Lv, Shuanggang Ma, Zhaodi Fu, Xiaoguang Chen, Shishan Yu, and Zeper Abliz
Analytical Chemistry 2013 Volume 85(Issue 5) pp:2977
Publication Date(Web):February 5, 2013
DOI:10.1021/ac400009s
Whole-body molecular imaging is able to directly map spatial distribution of molecules and monitor its biotransformation in intact biological tissue sections. Imaging mass spectrometry (IMS), a label-free molecular imaging method, can be used to image multiple molecules in a single measurement with high specificity. Herein, a novel easy-to-implement, whole-body IMS method was developed with air flow-assisted ionization in a desorption electrospray ionization mode. The developed IMS method can effectively image molecules in a large whole-body section in open air without sample pretreatment, such as chemical labeling, section division, or matrix deposition. Moreover, the signal levels were improved, and the spatial assignment errors were eliminated; thus, high-quality whole-body images were obtained. With this novel IMS method, in situ mapping analysis of molecules was performed in adult rat sections with picomolar sensitivity under ambient conditions, and the dynamic information of molecule distribution and its biotransformation was provided to uncover molecular events at the whole-animal level. A global view of the differential distribution of an anticancer agent and its metabolites was simultaneously acquired in whole-body rat and model mouse bearing neuroglioma along the administration time. The obtained drug distribution provided rich information for identifying the targeted organs and predicting possible tumor spectrum, pharmacological activity, and potential toxicity of drug candidates.
Co-reporter:Wei Yang, Yanhua Chen, Cong Xi, Ruiping Zhang, Yongmei Song, Qimin Zhan, Xiaofeng Bi, and Zeper Abliz
Analytical Chemistry 2013 Volume 85(Issue 5) pp:2606
Publication Date(Web):February 6, 2013
DOI:10.1021/ac303576b
Metabonomics is an important platform for investigating the metabolites of integrated living systems and their dynamic responses to changes caused by both endogenous and exogenous factors. A metabonomics strategy based on liquid chromatography–mass spectrometry/mass spectrometry in both positive and negative ion modes was applied to investigate the short-term and long-term stability of metabolites in plasma. Principal components analysis and ten types of identified metabolites were used to summarize the time-dependent change rules in metabolites systematically at different temperatures. The long-term stability of metabolites in plasma specimens stored at −80 °C for five years was also studied. Analysis of these subjects identified 36 metabolites with statistically significant changes in expression (p < 0.05) and found a kind of metabolite with a hundred-fold change. The stability of metabolites in blood at 4 °C for 24 h was also investigated. These studies show that a thorough understanding of the effects of metabolite stability are necessary for improving the reliability of potential biomarkers.
Co-reporter:Jiang Huang, Jianghao Sun, Yanhua Chen, Yongmei Song, Lijia Dong, Qinmin Zhan, Ruiping Zhang, Zeper Abliz
Analytica Chimica Acta 2012 Volume 711() pp:60-68
Publication Date(Web):20 January 2012
DOI:10.1016/j.aca.2011.10.058
A rapid, sensitive, specific and accurate analytical method of ultra-fast liquid chromatography combined with tandem mass spectrometry (UFLC–MS/MS) was established for simultaneous quantitative analysis of 16 distinct endogenous estrogens and their metabolites (EMs) in postmenopausal female urine. The quantitative method utilized a hydrolysis/extraction/derivatization step and a UFLC system to achieve separation in 16 min. The lower limit of quantitation for each estrogen metabolite was 2 pg mL−1 with the percent recovery of a known added amount of estrogen at 93.2–109.3%. The intra-batch accuracy and precision for all analytes were 87.5–107.7% and 0.6–11.7%, respectively, while inter-batch accuracy and precision were 87.0–105.8% and 1.2–10.2%, respectively. Using this developed and validated method, the comprehensive metabolic profiling of 16 EMs in urine samples of 86 postmenopausal female breast cancer patients and 36 healthy controls was investigated by systematic statistical analysis. As a result, the circulating levels of 6 EMs were found to be different by a comparison of patients and healthy controls. The parent estrogens, estrone (E1) and 17β-estradiol (E2), as well as 2-hydroxyestradiol (2-OHE2) and 4-hydroxyestradiol (4-OHE2) were produced in higher abundance, whereas 16α-hydroxyestrone (16α-OHE1) and 2-methoxyestradiol (2-MeOE2) were decreased in the breast cancer group. 2-OHE2 and 4-OHE2 in particular showed significant elevation in patients, which are consistent with the carcinogenic mechanism hypothesis that catechol estrogens can react with DNA via quinones, resulting in mutations to induce breast cancer. Thus, 2,4-hydroxylation may be the dominant metabolic pathway for parent estrogens rather than 16α-hydroxylation. The lower level of 2-MeOE2 in the breast cancer group was believed to correlate with its protective effect against tumor formation. This study could provide valuable information on the association of the EM metabolic pathway with carcinogenesis as well as identify potential biomarkers for estrogen-induced breast cancer risk.
Co-reporter:Yaping Tian, Jiuming He, Ruiping Zhang, Haining Lv, Shuanggang Ma, Yanhua Chen, Shishan Yu, Xiaoguang Chen, Yan Wu, Wenyi He, Zeper Abliz
Analytica Chimica Acta 2012 Volume 731() pp:60-67
Publication Date(Web):20 June 2012
DOI:10.1016/j.aca.2012.04.024
An integrated approach combining data acquisition using MSE and multi-period product ion scan (mpMS/MS), with high-resolution characteristic extracted ion chromatograms (hcXIC) as a data mining method, was developed for in vivo drug metabolites screening and identification. This approach is illustrated by analyzing metabolites of a potential anticancer agent, 3,6,7-trimethoxyphenanthroindolizidine (CAT) in rat urine based on rapid resolution liquid chromatography combined with tandem mass spectrometry (RRLC–MS/MS). Untargeted full-scan MSE enabled the high-throughput acquisition of potential metabolites, and targeted mpMS/MS contributed to the sensitivity and specificity of the acquisition of molecules of interest. The data processing method hcXIC, based on the structure of CAT, was shown to be highly effective for the metabolite discovery. Through the double-filtering effect of the characteristic ion and accurate mass, conventional extracted ion chromatograms that contained a substantial number of false-positive peaks were simplified into chromatograms essentially free of endogenous interferences. As a result, 21 metabolites were detected in rat urine after oral administration of CAT. Based on the characteristic fragmentation patterns of the phenanthroindolizidine alkaloid, the structures of 9 metabolites were identified. Furthermore, the interpretation of the MS/MS spectra of these metabolites enabled the determination of demethylation position as well as the differentiation between N-oxidized and hydroxylated metabolites.Graphical abstractHighlights► A RRLC–MS/MS approach was developed for metabolite analysis. ► MSE and mpMS/MS were integrated to improve throughput and sensitivity. ► The metabolites of a potential anticancer agent CAT were detected in rat urine. ► Demethylated, N-oxidized and hydroxylated metabolites were identified.
Co-reporter:Jing XU, Ya-Ping TIAN, Yan-Hua CHEN, Rui-Ping ZHANG, Fen YANG, Yong-Mei SONG, Abliz ZEPER
Chinese Journal of Analytical Chemistry 2011 Volume 39(Issue 12) pp:1793-1797
Publication Date(Web):December 2011
DOI:10.1016/S1872-2040(10)60487-2
Co-reporter:Dongmei Dai, Jiuming He, Ruixiang Sun, Ruiping Zhang, Haji Akber Aisa, Zeper Abliz
Analytica Chimica Acta 2009 Volume 632(Issue 2) pp:221-228
Publication Date(Web):26 January 2009
DOI:10.1016/j.aca.2008.11.002
NMR and LC–MS combined with an incompleted separation strategy were proposed to the simultaneous structure identification of natural products in crude extracts, and a novel method termed as NMR/LC–MS parallel dynamic spectroscopy (NMR/LC–MS PDS) was developed to discover the intrinsic correlation between retention time (Rt), mass/charge (m/z) and chemical shift (δ) data of the same constituent from mixture spectra by the co-analysis of parallelly visualized multispectroscopic datasets from LC–MS and 1H NMR. The extracted ion chromatogram (XIC) and 1H NMR signals deriving from the same individual constituent were correlated through fraction ranges and intensity changing profiles in NMR/LC–MS PDS spectrum due to the signal amplitude co-variation resulted from the concentration variation of constituents in a series of incompletely separated fractions. NMR/LC–MS PDS was applied to identify 12 constituents in an active herbal extract including flavonol glycosides, which was separated into a series of fractions by flash column chromatography. The complementary spectral information of the same individual constituent in the crude extract was discovered simultaneously from mixture spectra. Especially, two groups of co-eluted isomers were identified successfully. The results demonstrated that NMR/LC–MS PDS combined with the incompleted separation strategy achieved the similar function of on-line LC–NMR–MS analysis in off-line mode and had the potential for simplifying and accelerating the analytical routes for structure identification of constituents in herbs or their active extracts.
Co-reporter:Yanhua Chen, Ruiping Zhang, Yongmei Song, Jiuming He, Jianghao Sun, Jinfa Bai, Zhuoling An, Lijia Dong, Qimin Zhan and Zeper Abliz
Analyst 2009 vol. 134(Issue 10) pp:2003-2011
Publication Date(Web):14 Aug 2009
DOI:10.1039/B907243H
A metabonomics strategy based on rapid resolution liquid chromatography/tandem mass spectrometry (RRLC-MS/MS), multivariate statistics and metabolic correlation networks has been implemented to find biologically significant metabolite biomarkers in breast cancer. RRLC-MS/MS analysis by electrospray ionization (ESI) in both positive and negative ion modes was employed to investigate human urine samples. The resulting data matrices were analyzed using multivariate analysis. Application of orthogonal projections to latent structures discriminate analysis (OPLS-DA) allowed us to extract several discriminated metabolites reflecting metabolic characteristics between healthy volunteers and breast cancer patients. Correlation network analysis between these metabolites has been further applied to select more reliable biomarkers. Finally, high resolution MS and MS/MS analyses were performed for the identification of the metabolites of interest. We identified 12 metabolites as potential biomarkers including amino acids, organic acids, and nucleosides. They revealed elevated tryptophan and nucleoside metabolism as well as protein degradation in breast cancer patients. These studies demonstrate the advantages of integrating metabolic correlation networks with metabonomics for finding significant potential biomarkers: this strategy not only helps identify potential biomarkers, it also further confirms these biomarkers and can even provide biochemical insights into changes in breast cancer.
Co-reporter:Na Guo, Ruiping Zhang, Fei Song, Jiuming He, Bin Xia, Zeper Abliz
Journal of the American Society for Mass Spectrometry 2009 Volume 20(Issue 5) pp:845-851
Publication Date(Web):May 2009
DOI:10.1016/j.jasms.2008.12.024
Coldspray ionization (CSI) mass spectrometry, a variant of electrospray ionization (ESI) operating at low temperature (20 to −80 °C), has been used to characterize protein conformation and noncovalent complexes. A comparison of CSI and ESI was presented for the investigation of the equilibrium acid-induced unfolding of cytochrome c, ubiquitin, myoglobin, and cyclophilin A (CypA) over a wide range of pH values in aqueous solutions. CSI and nanoelectrospray ionization (nanoESI) were also compared in their performance to characterize the conformational changes of cytochrome c and myoglobin. Significant differences were observed, with narrower charged-state distribution and a shift to lower charge state in the CSI mass spectra compared with those in ESI and nanoESI mass spectra. The results suggest that CSI is more prone to preserving folded protein conformations in solution than the ESI and nanoESI methods. Moreover, the CSI-MS data are comparable with those obtained by other established biophysical methods, which are generally acknowledged to be the suitable techniques for monitoring protein conformation in solution. Noncovalent complexes of holomyoglobin and the protein–ligand complex between CypA and cyclosporin A (CsA) were also investigated at a neutral pH using the CSI-MS method. The results of this study suggest the ability of CSI-MS in retaining of protein conformation and noncovalent interactions in solution and probing subtle protein conformational changes. Additionally, the CSI-MS method is capable of analyzing quantitatively equilibrium unfolding transitions of proteins. CSI-MS may become one of the promising techniques for investigating protein conformation and noncovalent protein–ligand interactions in solution.A comparison of ESI, nanoESI, and CSI for characterizing the equilibrium acid-induced protein conformational changes and noncovalent complexes with mass spectrometry.Figure optionsDownload full-size imageDownload high-quality image (122 K)Download as PowerPoint slide
Co-reporter:Yue Liu, Jianbei Li, Shishan Yu, Zeper Abliz, Yunbao Liu, Jing Qu, Jing Liu, Youcai Hu
Analytica Chimica Acta 2008 Volume 611(Issue 2) pp:187-196
Publication Date(Web):24 March 2008
DOI:10.1016/j.aca.2008.01.076
The fragmentation behaviors of the two types of modified pregnane glycosides from Cynanchum forrestii were investigated by positive ion electrospray ionization multi-stage tandem mass spectrometry equipped with an ion trap analyzer. The spectral data further illuminated the predominance of ESI-MSn technique on the identification of pregnane glycosides, especially of two sorts of modified pregnane glycosides with aglycone skeletons of 13,14:14,15-disecopregnane-type and 14,15-secopregnane-type, which differed in the presence of the characteristic [M−46+Na]+ ion. For sugar residues, the fragment ions were analyzed and some possible fragmentation pathways were proposed, especially for 3-demethyl-2-deoxythevetose, the glycosidic cleavage reaction was easier to occur than those of other sugar units in its moiety. The natures and differences of the pregnane cores, and the types and linked sequences of sugar residues were illustrated. According to these conclusions, eight new pregnane glycosides in the fraction of Cynanchum forrestii were structurally elucidated by HPLC/HRMS and HPLC–DAD/ESI-MSn techniques. Results of the present studies can benefit the rapid identification and structural determination of analogous constituents in crude plant extracts.
Co-reporter:Jin-ping Qiao, Zeper Abliz, Feng-ming Chu, Pei-ling Hou, Li-yan Zhao, Min Xia, Yan Chang, Zong-ru Guo
Journal of Chromatography B 2004 Volume 805(Issue 1) pp:93-99
Publication Date(Web):5 June 2004
DOI:10.1016/j.jchromb.2004.02.017
6-Aminobutylphthalide (ABP) is a new drug candidate which is currently being developed for the treatment of cerebral ischemia. The pharmacokinetics and metabolism of ABP were studied using in situ microdialysis sampling in the brains of awake freely-moving rats. Two LC-MS/MS methods were used for the quantitative and qualitative analysis of microdialysate. For comparison and confirmation, brain tissue samples were also analyzed by LC-MS/MS and GC/MS. The results described provide more authentic information in pharmacokinetics and metabolism at the site of action by using the coupling of microdialysis to LC-MS/MS technique than the traditional sampling methods.

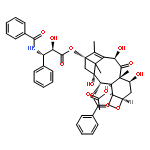
![Benzenepropanoic acid, b-(benzoylamino)-a-hydroxy-,(2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-6-(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-4,11,12b-trihydroxy-4a,8,13,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca[3,4]benz[1,2-b]oxet-9-ylester, (aR,bS)-](http://img.cochemist.com/ccimg/160600/160511-46-6.png)
![Benzenepropanoic acid, b-(benzoylamino)-a-hydroxy-,(2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-6-(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-4,11,12b-trihydroxy-4a,8,13,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca[3,4]benz[1,2-b]oxet-9-ylester, (aR,bS)-](http://img.cochemist.com/ccimg/160600/160511-46-6_b.png)
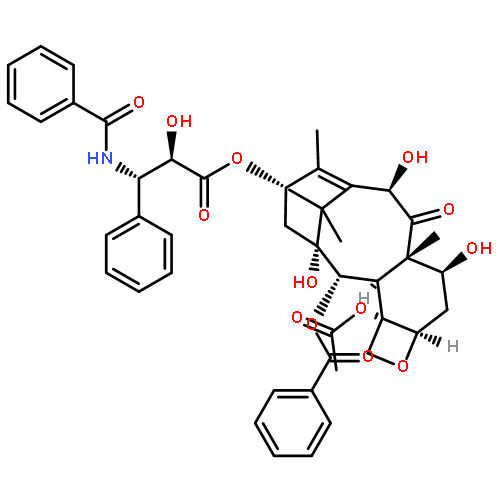
![Benzenepropanoic acid, b-(benzoylamino)-a,4-dihydroxy-,(2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-6,12b-bis(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-4,11-dihydroxy-4a,8,13,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca[3,4]benz[1,2-b]oxet-9-ylester, (aR,bS)-](http://img.cochemist.com/ccimg/132200/132160-32-8.png)
![Benzenepropanoic acid, b-(benzoylamino)-a,4-dihydroxy-,(2aR,4S,4aS,6R,9S,11S,12S,12aR,12bS)-6,12b-bis(acetyloxy)-12-(benzoyloxy)-2a,3,4,4a,5,6,9,10,11,12,12a,12b-dodecahydro-4,11-dihydroxy-4a,8,13,13-tetramethyl-5-oxo-7,11-methano-1H-cyclodeca[3,4]benz[1,2-b]oxet-9-ylester, (aR,bS)-](http://img.cochemist.com/ccimg/132200/132160-32-8_b.png)