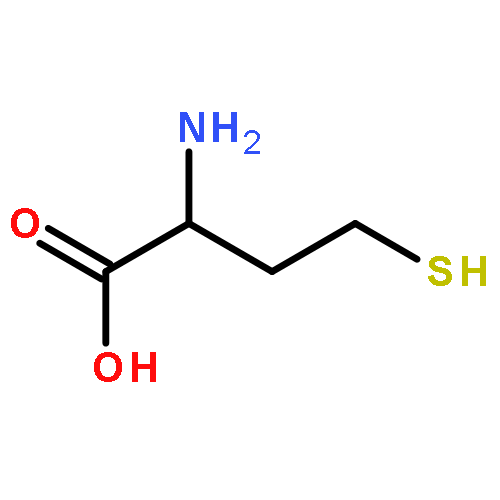
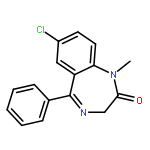
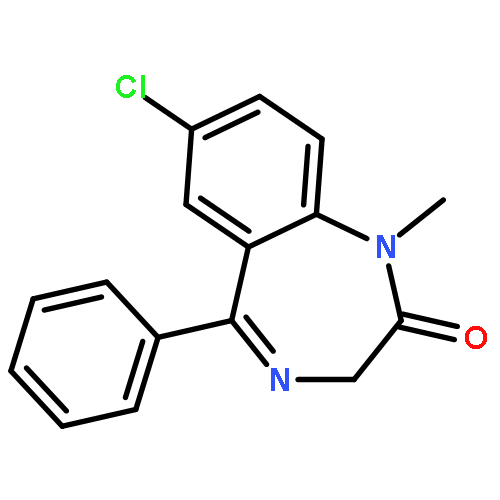
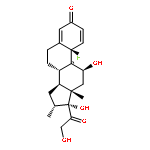
Co-reporter:Anyue Yin;Dewei Shang;Yuguan Wen;Liang Li
European Journal of Clinical Pharmacology 2016 Volume 72( Issue 8) pp:933-944
Publication Date(Web):2016 August
DOI:10.1007/s00228-016-2040-2
The aim of this study was to build an eligible population pharmacokinetic (PK) model for olanzapine in Chinese psychotic patients based on therapeutic drug monitoring (TDM) data, with assistance of meta-analysis, to facilitate individualized therapy.Population PK analysis for olanzapine was performed using NONMEM software (version 7.3.0). TDM data were collected from Guangzhou Brain Hospital (China). Because of the limitations of TDM data, model-based meta-analysis was performed to construct a structural model to assist the modeling of TDM data as prior estimates. After analyzing related covariates, a simulation was performed to predict concentrations for different types of patients under common dose regimens.A two-compartment model with first-order absorption and elimination was developed for olanzapine oral tablets, based on 23 articles with 390 data points. The model was then applied to the TDM data. Gender and smoking habits were found to be significant covariates that influence the clearance of olanzapine. To achieve a blood concentration of 20 ng/mL (the lower boundary of the recommended therapeutic range), simulation results indicated that the dose regimen of olanzapine should be 5 mg BID (twice a day), ≥ 5 mg QD (every day) plus 10 mg QN (every night), or >10 mg BID for female nonsmokers, male nonsmokers and male smokers, respectively.The population PK model, built using meta-analysis, could facilitate the modeling of TDM data collected from Chinese psychotic patients. The factors that significantly influence olanzapine disposition were determined and the final model could be used for individualized treatment.
Co-reporter:Yu-Peng Ren;Ru-Jia Xie;Scott Marshall
European Journal of Clinical Pharmacology 2015 Volume 71( Issue 10) pp:1209-1221
Publication Date(Web):2015 October
DOI:10.1007/s00228-015-1918-8
To quantify pharmacokinetic (PK) and pharmacodynamic (PD) relationships of various classes of GABAA agonists in healthy volunteers, in order to investigate the sensitivity of the biomarker responses due to differing GABAA-subtype selectivity and to explore the correlation between biomarker responses and side effects of these drugs.A comprehensive search was conducted for published placebo-controlled clinical studies of non- and α1-selective GABAA drugs in healthy volunteers. PK/PD models were developed for concentrations and biomarker outcomes (saccadic eye movement (SEM), visual analogue scale (VAS), digit symbol substitution task (DSST), and critical flicker fusion test (CFFT)) extracted from included studies. Predicted responses and equivalent doses for biomarkers (based on predicted response) were used to compare drug effects. And the relationship between biomarkers and safety was explored by linear regression.A total of 2237 data from 163 articles were included. Based on PK and placebo effect modeling, linear biomarker-concentration relationships well fit the data. The α1-selective compounds had similar equivalent doses for VAS, DSST, and CFFT (4.7–6.7 mg), which were about three to seven times lower than that for SEM (14.4–35.5 mg), while such difference was less evident for non-selective drugs. DSST had the highest correlations with incidences of somnolence and dizziness.The integral PK/PD models of GABAA agonists were established in healthy volunteers. SEM was identified as the most sensitive biomarker in differentiating GABAA receptor α1 subtype selective compounds. The exploratory analysis implied that different relationships existed between the drug effects on biomarkers and the adverse event profiles in healthy volunteers.
Co-reporter:Qinghong Su, Jian Li, Xiwei Ji, Jingyun Li, Tianyan Zhou, Wei Lu, Liang Li
Journal of Chromatography B 2015 Volume 985() pp:119-123
Publication Date(Web):15 March 2015
DOI:10.1016/j.jchromb.2015.01.024
•An LC-MS/MS method for the determination of cabozantinib in rat plasma.•This method was fully validated and successfully applied to a pharmacokinetic study.•The LLOQ for cabozantinib in this method is 0.5 ng/mL, which is much lower than the literature.•The elimination half-life of cabozantinib in rat is 4.91 h.A simple, rapid and sensitive high-performance liquid chromatography tandem mass-spectrometric method (LC-MS/MS) for the novel multiple tyrosine kinase inhibitor (TKI) cabozantinib was developed and validated using erlotinib as internal standard (IS). Plasma samples were pre-treated by liquid–liquid extraction with ethyl acetate. Separation was achieved on a reversed phase C18 column (50 × 2 mm, 5 μm) at ambient temperature using isocratic elution with acetonitrile-water (45:55, v/v) containing 5 mM ammonium formate buffer (finally adjusted to apparent pH* = 5.0 with formic acid) at a flow rate of 0.4 mL/min. The analytes were monitored by a triple quadrupole tandem mass spectrometer with electrospray ionization source in the positive ion mode. Calibration curve was linear (r > 0.99) in a concentration range of 0.5–1000 ng/mL with the lower limit of quantification (LLOQ) of 0.5 ng/mL. Intra- and inter-day accuracy and precision were validated by relative error values (RE) and relative standard deviation values (RSD), respectively, which were both lower than the limit of 15%. This method was successfully applied to a pharmacokinetic study of cabozantinib in rats.
Co-reporter:Mengyao Li;Hanqing Li;Xiaoliang Cheng;Xipei Wang;Liang Li
Pharmaceutical Research 2013 Volume 30( Issue 5) pp:1400-1408
Publication Date(Web):2013 May
DOI:10.1007/s11095-013-0978-7
To investigate the pharmacological effects of different erlotinib (ER) and gemcitabine (GM) combination schedules by in vitro and in vivo experiments and PK/PD models in non-small cell lung cancer cells.H1299 cells were exposed to different ER combined with GM schedules. Cell growth inhibition was analyzed to evaluate these schedules. A preclinical in vivo study was then conducted to compare tumor suppression effects of different schedules in H1299 xenografts. PK/PD models were developed to quantify the anti-tumor interaction of ER and GM.Synergism was observed when ER preceded GM, but other sequences showed antagonism. The optimal in vitro schedule, or interval schedule, was applied to the animal study, which showed greater anti-tumor effect than simultaneous group. PK/PD models implied that interaction of the two drugs was additive in simultaneous treatment but synergistic in interval schedule. The simulation results showed that interval schedule can delay tumor growth for a longer time, and demonstrated more evident anti-tumor effect compared with simultaneous group if the treatment duration was longer.Interval schedule of the two drugs can achieve synergistic anti-tumor effect, and is superior to simultaneous treatment.
Co-reporter:Xiaoliang Cheng, Liping Guo, Zaiquan Li, Liang Li, Tianyan Zhou, Wei Lu
Journal of Chromatography B 2013 Volumes 915–916() pp:64-70
Publication Date(Web):1 February 2013
DOI:10.1016/j.jchromb.2012.12.020
A HPLC method with on-line solid phase extraction (SPE) and column switching was developed for simultaneous determination of 5-aminoimidazole-4-carboxamide riboside (AICA riboside) and its active metabolite 5-aminoimidazole-4-carboxamide ribotide (AICA ribotide) in nude mice plasma. Plasma sample was deproteinized by adding a half volume of 10% trichloroacetic acid (TCA), and the resulting supernatant was extracted with diethyl ether to remove TCA. 50 μl aqueous fraction was injected onto a WAX-1 SPE column, and AICA ribotide was trapped on the SPE column, while AICA riboside was eluted from the SPE column. The chromatographic separation of AICA riboside was achieved on CG16 column, and separation of AICA ribotide was performed on HILIC-10 and WAX-1 column. The columns temperature was maintained at 40 °C, and the optimal detection wavelength was 268 nm for both AICA riboside and AICA ribotide. The total analytical run time was 40 min. The proposed method was linear over the range of 0.1–500 μg/ml for AICA riboside and 0.03–50 μg/ml for AICA ribotide. The lower limit of quantification (LLOQ) was 100 and 30 ng/ml for AICA riboside and AICA ribotide, respectively. The sensitivity, accuracy and precision of this method were within acceptable limits during validation period. The method was successfully applied to investigate the pharmacokinetics characteristics of AICA riboside and its active metabolite AICA ribotide in nude mice bearing MCF-7 cell xenografts.Highlights► A HPLC method for simultaneous assay of AICA riboside and AICA ribotide was developed. ► This HPLC method involved on-line solid phase extraction and column switching. ► The method was applied to pharmacokinetics study of AICA riboside and AICA ribotide.
Co-reporter:Xingang Li;Liang Li;Xipei Wang;Yupeng Ren;Tianyan Zhou
Journal of Pharmaceutical Sciences 2012 Volume 101( Issue 10) pp:3946-3961
Publication Date(Web):
DOI:10.1002/jps.23236
Abstract
The objective of this study was to characterize exenatide double-walled microspheres (DWMS) using model-based methods. Exenatide DWMS were prepared using oil-in-oil-in-water method, and physicochemical characterization and in vitro release and degradation of DWMS were evaluated. The pharmacokinetics (PK) and pharmacodynamics (PD) were investigated after subcutaneous injection to diabetic rats. Transit compartment model was used to describe the in vivo release of exenatide from DWMS successfully. On the basis of the insulinotropic effects of exenatide and hypoglycemic effects of insulin, PK/PD model was developed and nicely described the concentration–effect relationship of exenatide. Moreover, on the basis of the transit compartment model, a simulation method was applied to predict in vivo release, and in vitro and in vivo correlation was established. In conclusion, DWMS was a promising vehicle for delivery of exenatide, and the proposed PK/PD model allowed a better understanding of the pharmacological properties of exenatide DWMS. Transit compartment model-based modeling and simulation methods provided more options for the description and prediction of the in vivo exenatide release from DWMS. © 2012 Wiley Periodicals, Inc. and the American Pharmacists Association J Pharm Sci 101:3946–3961, 2012
Co-reporter:Jiaojiao Xu, Ye Chen, Liang Li, Zaiquan Li, Chenai Wang, Tianyan Zhou, Wei Lu
Journal of Pharmaceutical and Biomedical Analysis 2012 Volume 62() pp:182-186
Publication Date(Web):25 March 2012
DOI:10.1016/j.jpba.2011.12.015
Sulfotransferase-catalyzed (SULT-catalyzed) sulfation is responsible for hormone regulation and xenobiotic detoxification. In the current study, a sensitive and widely applicable method of reversed-phase HPLC–UV was developed for the determination of 3′-phosphoadenosine 5′-phosphate (PAP), a common product of different subtypes of sulfation reactions, in HepG2 cells. The analyte was separated on a ZORBAX Extend-C18 column with the mobile phase of methanol and water containing 75 mM KH2PO4, 100 mM NH4Cl, and 1 mM 1-octylamine (pH 4.55), at a flow rate of 1.0 ml/min. The assay exhibited linearity over the range of 0.1–20 μM for PAP with a correlation coefficient of 0.9995. The total time per run was under 10 min. The intra- and inter-day precision was less than 7.2%, with accuracy in the range 82.6–102.0%. The method was applied to the determination of kinetic parameters Km, Vm, and Kcat, of three different SULTs (SULT1A1, SULT2A1, and SULT1E1) in HepG2. This universal HPLC–UV method was easy to perform, economically feasible, and suitably efficient for the investigation of the enzyme kinetics of the SULT family using multiplex substrates.Highlights► A quantitative method for PAP, a common product of sulfation reactions, in HepG2 cells was developed and fully validated. ► Theophylline as internal standard allowed relative quantification. ► The method was applied to the determination of kinetic parameters of three different SULT subtypes in HepG2 cells.
Co-reporter:Xipei Wang;Dewei Shang;Jakob Ribbing
European Journal of Clinical Pharmacology 2012 Volume 68( Issue 8) pp:1157-1166
Publication Date(Web):2012 August
DOI:10.1007/s00228-012-1245-2
Our objective was to describe the time course of the placebo effect in asthma and quantitatively investigate the affective factors of the placebo effect for the placebo response simulation during the asthma clinical study design.We conducted a systemic search of public data sources for the study-level forced expiratory volume in 1 second (FEV1) to build the placebo effect model for studies by oral or inhaled administrations simultaneously. The administration routes, types of inhalation device, mean patient age, mean male proportion, baseline FEV1, disease severity, year of publication, inhaled corticosteroid status during the treatment, and dropout rate were tested as covariates.There are 34 literature sources containing 178 mean values for FEV1 presenting the individual observations from about 3,703 patients. The exponential models adequately described the time course of placebo effect with the typical value of the maximum placebo effect (Pmax) of 0.060 L. Dropout rate incorporated in the residual error model and the disease severity (mild to moderate and moderate to severe) at baseline were covariates that remained in the final model.The placebo effect is adequately described by an exponential model over time. By incorporating the dropout rate in the residual error model, the estimation precision was improved. The model could predict the placebo response profile in mild to severe asthmatic patients for the asthma clinical study design and could also be a structure model of the placebo effect for the pure drug effect evaluation in the asthma clinical trials.
Co-reporter:Hanqing Li;Mengyao Li;Zaiquan Li;Liang Li;Shanshan Bi;Chenhui Deng;Rui Chen;Tianyan Zhou
Chinese Journal of Chemistry 2011 Volume 29( Issue 8) pp:1709-1714
Publication Date(Web):
DOI:10.1002/cjoc.201180305
Abstract
The synthesis and differential antiproliferative activity of two active isomeric metabolites of erlotinib were investigated. This synthetic process had demonstrated to avoid the unstable 4-chloroquinazoline intermediates and long procedures. New intermediates and final compounds were identified by 1H NMR, 13C NMR and ESI-TOF MS, and their purities were determined by high performance liquid chromatography. In vitroproliferative assay indicated that these two metabolites possessed antiproliferative activity against some conventional tumor cell lines and EGFR tyrosine kinase over-expression tumor cell lines as compared to erlotinib control, and their antitumor activity in cellular level was first reported here.
Co-reporter:Dewei Shang, Xipei Wang, Xiutai Zhao, Fengming Huang, Ganzhong Tian, Wei Lu, Tianyan Zhou
Journal of Chromatography B 2011 Volume 879(Issue 30) pp:3459-3464
Publication Date(Web):15 November 2011
DOI:10.1016/j.jchromb.2011.09.025
A HPLC method with on-line solid phase extraction (SPE) and DAD detection was developed for the simultaneous determination of nitrendipine and hydrochlorothiazide in spontaneously hypertensive rat (SHR) plasma. Plasma samples (100 μL) were injected directly onto a CAPCELL MF C8 SPE column. High-abundance proteins and most matrixes in plasma were removed by on-line SPE technology, while nitrendipine and hydrochlorothiazide trapped on the SPE column were effectively separated on a C18 analytical column. The column temperature was maintained at 20 °C. The optimal detection wavelength was 237 nm for NTDP and 271 nm for HCTZ. The total analytical run time was 34 min. The proposed method was linear over the range 5–500 ng mL−1 for nitrendipine and 10–1000 ng mL−1 for hydrochlorothiazide. The lower limit of detection (LLOD) was 0.5 and 0.6 ng mL−1 for nitrendipine and hydrochlorothiazide, respectively. The sensitivity and precision of the method were within acceptable limits during validation period. The method was successfully used to investigate the pharmacokinetic characteristics of nitrendipine and hydrochlorothiazide in spontaneously hypertensive rats.Highlights► Simultaneous determination of two chemical compounds with different polarity and solubility (NTDP and HCTZ) in SHR plasma using HPLC with on-line solid-phase extraction. ► Many anti-hypertension drugs were lipophilic, and the polarity and solubility was different from HCTZ. So this method may be used for the simultaneous quantification in combination therapy with HCTZ. ► The method is simple, sensitive and reliable.
Co-reporter:Xingang Li, Liang Li, Xipei Wang, Yupeng Ren, ... Wei Lu
Journal of Pharmaceutical Sciences (October 2012) Volume 101(Issue 10) pp:3946-3961
Publication Date(Web):1 October 2012
DOI:10.1002/jps.23236
The objective of this study was to characterize exenatide double‐walled microspheres (DWMS) using model‐based methods. Exenatide DWMS were prepared using oil‐in‐oil‐in‐water method, and physicochemical characterization and in vitro release and degradation of DWMS were evaluated. The pharmacokinetics (PK) and pharmacodynamics (PD) were investigated after subcutaneous injection to diabetic rats. Transit compartment model was used to describe the in vivo release of exenatide from DWMS successfully. On the basis of the insulinotropic effects of exenatide and hypoglycemic effects of insulin, PK/PD model was developed and nicely described the concentration–effect relationship of exenatide. Moreover, on the basis of the transit compartment model, a simulation method was applied to predict in vivo release, and in vitro and in vivo correlation was established. In conclusion, DWMS was a promising vehicle for delivery of exenatide, and the proposed PK/PD model allowed a better understanding of the pharmacological properties of exenatide DWMS. Transit compartment model‐based modeling and simulation methods provided more options for the description and prediction of the in vivo exenatide release from DWMS.
Co-reporter:Xuan Zhou, Dewei Shang, Tianlan Zhang, Liang Li, Tianyan Zhou, Wei Lu
Pharmacological Research (August 2012) Volume 66(Issue 2) pp:177-184
Publication Date(Web):1 August 2012
DOI:10.1016/j.phrs.2012.04.001
Angiotensin II (Ang II) and angiotensin-(1-7) (Ang-(1-7)) are biologically active effectors in the renin–angiotensin system (RAS) and have been demonstrated to have potential function in predicting cardiovascular diseases. We developed mechanism-based mathematical models to characterize the up/down-regulation of Ang II/Ang-(1-7), and the effects of perindopril on hypertension progression in spontaneously hypertensive rats (SHR). SHR were randomly assigned to the control group (n = 6) and treatment group (n = 6). Rats in the treatment group received oral perindopril (5 mg kg−1 day−1). Systolic blood pressure (SBP) was measured by the tail-cuff method. Serum Ang II and Ang-(1-7) concentrations were determined by enzyme-linked immunosorbent assay (ELISA). Three linked turnover models were developed to describe Ang II, Ang-(1-7) and SBP profiles. All parameters were estimated using nonlinear mixed-effects modeling. The results showed that Ang II, Ang-(1-7) and SBP gradually increased in the control group. These counterbalance mechanisms were reflected in the models with two feedback cycles. It was assumed that the Ang-(1-7) production rate constant (Kin_Ang17) was stimulated by Ang II, and the Ang II output rate constant (Kout_Ang2) reflecting Ang II degradation was stimulated by Ang-(1-7). The decrease in Ang II and increase in Ang-(1-7) were observed in rats treated with perindopril. The models described the counterbalance relationship of Ang II and Ang-(1-7) well, and provided insights into ACE inhibition using perindopril. The models could be extended to incorporate other biomarkers and the effects of various ACE inhibitors (ACEIs).Download high-res image (124KB)Download full-size image
Co-reporter:Miaoran Ning, Liang Li, Jian Li, Zaiquan Li, Runtao Li, Tianyan Zhou, Wei Lu
Acta Pharmaceutica Sinica B (April 2012) Volume 2(Issue 2) pp:181-187
Publication Date(Web):April 2012
DOI:10.1016/j.apsb.2012.02.006