Co-reporter:Glen M. Hocky, Thomas Dannenhoffer-Lafage, and Gregory A. Voth
Journal of Chemical Theory and Computation September 12, 2017 Volume 13(Issue 9) pp:4593-4593
Publication Date(Web):August 11, 2017
DOI:10.1021/acs.jctc.7b00690
Many free-energy sampling and quantum mechanics/molecular mechanics (QM/MM) computations on protein complexes have been performed where, by necessity, a single component is studied isolated in solution while its overall configuration is kept in the complex-like state by either rigid restraints or harmonic constraints. A drawback in these studies is that the system’s native fluctuations are lost, both due to the change of environment and the imposition of the extra potential. Yet, we know that having both accurate structure and fluctuations is likely crucial to achieving correct simulation estimates for the subsystem within its native larger protein complex context. In this work, we provide a new approach to this problem by drawing on ideas developed to incorporate experimental information into a molecular simulation by relative entropy minimization to a target system. We show that by using linear biases on coarse-grained (CG) observables (such as distances or angles between large subdomains within a protein), we can maintain the protein in a particular target conformation while also preserving the correct equilibrium fluctuations of the subsystem within its larger biomolecular complex. As an application, we demonstrate this algorithm by training a bias that causes an actin monomer (and trimer) in solution to sample the same average structure and fluctuations as if it were embedded within a much larger actin filament. Additionally, we have developed a number of algorithmic improvements that accelerate convergence of the on-the-fly relative entropy minimization algorithms for this type of application. Finally, we have contributed these methods to the PLUMED open source free energy sampling software library.
Co-reporter:Gregory A. Voth
Accounts of Chemical Research March 21, 2017 Volume 50(Issue 3) pp:594-594
Publication Date(Web):March 21, 2017
DOI:10.1021/acs.accounts.6b00572
This Commentary will describe the goal of developing and implementing novel, powerful, and integrated multiscale computer simulation methodology capable of accessing the large length and long time scales inherent in the behavior of biomolecular, multiprotein “active matter” complexes within the context of cellular biology. Examples include those involved in the actin-based cytoskeleton and its mechanochemistry. The primary objective is to connect detailed molecular and chemical properties with the key mesoscopic features manifest at the scales of cellular biology through a transformative theoretical and computer simulation approach, based on real physical and chemical interactions. This multiscale computational work would also make critical contact with rapidly developing experimental techniques such as super-resolution optical imaging, single molecule spectroscopy, and cryo-electron tomography, which are providing remarkable insight into the internal mesoscale self-organization and dynamics of cells.
Co-reporter:James F. Dama, Jaehyeok Jin, and Gregory A. Voth
Journal of Chemical Theory and Computation March 14, 2017 Volume 13(Issue 3) pp:1010-1010
Publication Date(Web):January 23, 2017
DOI:10.1021/acs.jctc.6b01081
When viewed through a coarse-grained lens, important molecular and biophysical systems can appear to undergo discrete, switch-like state changes in addition to more continuous configurational motions. One of our recent papers described a theory for bottom-up coarse-graining of the equilibrium statistics of models with such behavior, called ultra-coarse-grained (UCG) models, and a follow up paper described an implementation when the states of the coarse-grained sites or “beads” change rarely. However, not all systems with this discrete behavior fall under that special limit. This article develops the general UCG theory for the opposite limit, that is, where the internal states of the CG particles or beads adjust rapidly so as to always remain effectively at quasi-equilibrium no matter what the positions of the coarse-grained particles. This rapid local equilibrium allows ultra-coarse-graining to mix standard coarse-grained force fields by using local order parameters to control the degree of mixing, which adds an environmental dependence and many-body effects to the coarse-grained model while requiring minimal new coding. This article first presents the definition of such UCG force fields as well as their fitting procedures from atomistic-scale data, and then it presents three examples of UCG simulations with an approach that we call UCG with rapid local equilibrium (UCG-RLE). We then present an application of UCG-RLE using the full bottom-up methodology to coarse-grain and simulate cooperative hydrophobic association of neopentane in methanol solvent. UCG-RLE force matching does a superior job of matching solute–solute correlation functions and solute cluster size distributions compared to the more standard force-matched models not having coarse-grained sites with discrete internal states.
Co-reporter:Mijo Simunovic, Anđela Šarić, J. Michael Henderson, Ka Yee C. Lee, and Gregory A. Voth
ACS Central Science December 27, 2017 Volume 3(Issue 12) pp:1246-1246
Publication Date(Web):November 21, 2017
DOI:10.1021/acscentsci.7b00392
Biological membranes have a central role in mediating the organization of membrane-curving proteins, a dynamic process that has proven to be challenging to probe experimentally. Using atomic force microscopy, we capture the hierarchically organized assemblies of Bin/amphiphysin/Rvs (BAR) proteins on supported lipid membranes. Their structure reveals distinct long linear aggregates of proteins, regularly spaced by up to 300 nm. Employing accurate free-energy calculations from large-scale coarse-grained computer simulations, we found that the membrane mediates the interaction among protein filaments as a combination of short- and long-ranged interactions. The long-ranged component acts at strikingly long distances, giving rise to a variety of micron-sized ordered patterns. This mechanism may contribute to the long-ranged spatiotemporal control of membrane remodeling by proteins in the cell.
Co-reporter:Rajib Biswas;Gregory A Voth
Journal of Chemical Sciences 2017 Volume 129( Issue 7) pp:1045-1051
Publication Date(Web):05 June 2017
DOI:10.1007/s12039-017-1283-5
The classic Marcus electron transfer reaction model demonstrated that a barrierless electron transfer reaction can occur when both the reactant and product have almost similar solvation environment. In our recently developed proton model, we have incorporated the pre-solvation concept and showed that it indeed facilitates the proton diffusion in aqueous solution. In this work, we further quantify the degree of pre-solvation using different structural parameters, e.g., tetrahedral order parameter, average numbers of hydrogen bonds. All the above said parameters exhibit a very strong correlation with the proton share parameter. The more Zundel-like configurations have almost identical solvation environment for both the water molecules and support the pre-solvation concept. However, in the case of Eigen-like configurations, the central hydronium and “special pair” water have distinctly different solvation structures.
Co-reporter:Christopher Arntsen, Chen Chen, Gregory A. Voth
Chemical Physics Letters 2017 Volume 683(Volume 683) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.cplett.2017.04.064
•Multiscale reactive molecular dynamics (MS-RMD) models at developed using relative entropy minimization.•Fitted using limited bulk phase ab initio molecular dynamics.•Two MS-RMD models are presented, with and without 3-body interaction potentials.•The model with 3-body terms faithfully reproduces solvation structures and proton transfer energetics.We present two new multiscale molecular dynamics (MS-RMD) models for the hydrated excess proton in water developed directly from ab initio molecular dynamics (AIMD) simulation data of the same system. The potential of mean force along the proton transfer reaction coordinate and radial distribution functions for the MS-RMD models are shown to faithfully reproduce those of AIMD. The models are developed using an algorithm based on relative entropy minimization, thus demonstrating the ability of the method to rapidly generate accurate and highly efficient reactive MD force fields.Download high-res image (94KB)Download full-size image
Co-reporter:Jesper J. MadsenAnton V. Sinitskiy, Jianing Li, Gregory A. Voth
Journal of Chemical Theory and Computation 2017 Volume 13(Issue 2) pp:
Publication Date(Web):January 2, 2017
DOI:10.1021/acs.jctc.6b01076
Numerous biomolecules and biomolecular complexes, including transmembrane proteins (TMPs), are symmetric or at least have approximate symmetries. Highly coarse-grained models of such biomolecules, aiming at capturing the essential structural and dynamical properties on resolution levels coarser than the residue scale, must preserve the underlying symmetry. However, making these models obey the correct physics is in general not straightforward, especially at the highly coarse-grained resolution where multiple (∼3–30 in the current study) amino acid residues are represented by a single coarse-grained site. In this paper, we propose a simple and fast method of coarse-graining TMPs obeying this condition. The procedure involves partitioning transmembrane domains into contiguous segments of equal length along the primary sequence. For the coarsest (lowest-resolution) mappings, it turns out to be most important to satisfy the symmetry in a coarse-grained model. As the resolution is increased to capture more detail, however, it becomes gradually more important to match modular repeats in the secondary structure (such as helix-loop repeats) instead. A set of eight TMPs of various complexity, functionality, structural topology, and internal symmetry, representing different classes of TMPs (ion channels, transporters, receptors, adhesion, and invasion proteins), has been examined. The present approach can be generalized to other systems possessing exact or approximate symmetry, allowing for reliable and fast creation of multiscale, highly coarse-grained mappings of large biomolecular assemblies.
Co-reporter:Sangyun Lee, Heather B. Mayes, Jessica M. J. Swanson, and Gregory A. Voth
Journal of the American Chemical Society 2016 Volume 138(Issue 45) pp:14923-14930
Publication Date(Web):October 26, 2016
DOI:10.1021/jacs.6b06683
The ClC family of transmembrane proteins functions throughout nature to control the transport of Cl– ions across biological membranes. ClC-ec1 from Escherichia coli is an antiporter, coupling the transport of Cl– and H+ ions in opposite directions and driven by the concentration gradients of the ions. Despite keen interest in this protein, the molecular mechanism of the Cl–/H+ coupling has not been fully elucidated. Here, we have used multiscale simulation to help identify the essential mechanism of the Cl–/H+ coupling. We find that the highest barrier for proton transport (PT) from the intra- to extracellular solution is attributable to a chemical reaction, the deprotonation of glutamic acid 148 (E148). This barrier is significantly reduced by the binding of Cl– in the “central” site (Cl–cen), which displaces E148 and thereby facilitates its deprotonation. Conversely, in the absence of Cl–cen E148 favors the “down” conformation, which results in a much higher cumulative rotation and deprotonation barrier that effectively blocks PT to the extracellular solution. Thus, the rotation of E148 plays a critical role in defining the Cl–/H+ coupling. As a control, we have also simulated PT in the ClC-ec1 E148A mutant to further understand the role of this residue. Replacement with a non-protonatable residue greatly increases the free energy barrier for PT from E203 to the extracellular solution, explaining the experimental result that PT in E148A is blocked whether or not Cl–cen is present. The results presented here suggest both how a chemical reaction can control the rate of PT and also how it can provide a mechanism for a coupling of the two ion transport processes.
Co-reporter:Rui Sun, James F. Dama, Jeffrey S. Tan, John P. Rose, and Gregory A. Voth
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 10) pp:5157-5169
Publication Date(Web):September 6, 2016
DOI:10.1021/acs.jctc.6b00206
Metadynamics is an important enhanced sampling technique in molecular dynamics simulation to efficiently explore potential energy surfaces. The recently developed transition-tempered metadynamics (TTMetaD) has been proven to converge asymptotically without sacrificing exploration of the collective variable space in the early stages of simulations, unlike other convergent metadynamics (MetaD) methods. We have applied TTMetaD to study the permeation of drug-like molecules through a lipid bilayer to further investigate the usefulness of this method as applied to problems of relevance to medicinal chemistry. First, ethanol permeation through a lipid bilayer was studied to compare TTMetaD with nontempered metadynamics and well-tempered metadynamics. The bias energies computed from various metadynamics simulations were compared to the potential of mean force calculated from umbrella sampling. Though all of the MetaD simulations agree with one another asymptotically, TTMetaD is able to predict the most accurate and reliable estimate of the potential of mean force for permeation in the early stages of the simulations and is robust to the choice of required additional parameters. We also show that using multiple randomly initialized replicas allows convergence analysis and also provides an efficient means to converge the simulations in shorter wall times and, more unexpectedly, in shorter CPU times; splitting the CPU time between multiple replicas appears to lead to less overall error. After validating the method, we studied the permeation of a more complicated drug-like molecule, trimethoprim. Three sets of TTMetaD simulations with different choices of collective variables were carried out, and all converged within feasible simulation time. The minimum free energy paths showed that TTMetaD was able to predict almost identical permeation mechanisms in each case despite significantly different definitions of collective variables.
Co-reporter:Thomas Dannenhoffer-Lafage, Andrew D. White, and Gregory A. Voth
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 5) pp:2144-2153
Publication Date(Web):April 5, 2016
DOI:10.1021/acs.jctc.6b00043
To extract meaningful data from molecular simulations, it is necessary to incorporate new experimental observations as they become available. Recently, a new method was developed for incorporating experimental observations into molecular simulations, called experiment directed simulation (EDS), which utilizes a maximum entropy argument to bias an existing model to agree with experimental observations while changing the original model by a minimal amount. However, there is no discussion in the literature of whether or not the minimal bias systematically and generally improves the model by creating agreement with the experiment. In this work, we show that the relative entropy of the biased system with respect to an ideal target is always reduced by the application of a minimal bias, such as the one utilized by EDS. Using all-atom simulations that have been biased with EDS, one can then easily and rapidly improve a bottom-up multiscale coarse-grained (MS-CG) model without the need for a time-consuming reparametrization of the underlying atomistic force field. Furthermore, the improvement given by the many-body interactions introduced by the EDS bias can be maintained after being projected down to effective two-body MS-CG interactions. The result of this analysis is a new paradigm in coarse-grained modeling and simulation in which the “bottom-up” and “top-down” approaches are combined within a single, rigorous formalism based on statistical mechanics. The utility of building the resulting EDS-MS-CG models is demonstrated on two molecular systems: liquid methanol and ethylene carbonate.
Co-reporter:Sangyun Lee, Ruibin Liang, Gregory A. Voth, and Jessica M. J. Swanson
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 2) pp:879-891
Publication Date(Web):January 6, 2016
DOI:10.1021/acs.jctc.5b01109
An important challenge in the simulation of biomolecular systems is a quantitative description of the protonation and deprotonation process of amino acid residues. Despite the seeming simplicity of adding or removing a positively charged hydrogen nucleus, simulating the actual protonation/deprotonation process is inherently difficult. It requires both the explicit treatment of the excess proton, including its charge defect delocalization and Grotthuss shuttling through inhomogeneous moieties (water and amino residues), and extensive sampling of coupled condensed phase motions. In a recent paper ( J. Chem. Theory Comput. 2014, 10, 2729−2737), a multiscale approach was developed to map high-level quantum mechanics/molecular mechanics (QM/MM) data into a multiscale reactive molecular dynamics (MS-RMD) model in order to describe amino acid deprotonation in bulk water. In this article, we extend the fitting approach (called FitRMD) to create MS-RMD models for ionizable amino acids within proteins. The resulting models are shown to faithfully reproduce the free energy profiles of the reference QM/MM Hamiltonian for PT inside an example protein, the ClC-ec1 H+/Cl– antiporter. Moreover, we show that the resulting MS-RMD models are computationally efficient enough to then characterize more complex 2-dimensional free energy surfaces due to slow degrees of freedom such as water hydration of internal protein cavities that can be inherently coupled to the excess proton charge translocation. The FitRMD method is thus shown to be an effective way to map ab initio level accuracy into a much more computationally efficient reactive MD method in order to explicitly simulate and quantitatively describe amino acid protonation/deprotonation in proteins.
Co-reporter:Ruibin Liang;Jessica M. J. Swanson;Yuxing Peng;Mårten Wikström
PNAS 2016 Volume 113 (Issue 27 ) pp:7420-7425
Publication Date(Web):2016-07-05
DOI:10.1073/pnas.1601982113
Cytochrome c oxidase (CcO) reduces oxygen to water and uses the released free energy to pump protons across the membrane. We have used multiscale
reactive molecular dynamics simulations to explicitly characterize (with free-energy profiles and calculated rates) the internal
proton transport events that enable proton pumping during first steps of oxidation of the fully reduced enzyme. Our results
show that proton transport from amino acid residue E286 to both the pump loading site (PLS) and to the binuclear center (BNC)
are thermodynamically driven by electron transfer from heme a to the BNC, but that the former (i.e., pumping) is kinetically favored whereas the latter (i.e., transfer of the chemical
proton) is rate-limiting. The calculated rates agree with experimental measurements. The backflow of the pumped proton from
the PLS to E286 and from E286 to the inside of the membrane is prevented by large free-energy barriers for the backflow reactions.
Proton transport from E286 to the PLS through the hydrophobic cavity and from D132 to E286 through the D-channel are found
to be strongly coupled to dynamical hydration changes in the corresponding pathways and, importantly, vice versa.
Co-reporter:Ruibin Liang;Jessica M. J. Swanson;Jesper J. Madsen;Mei Hong;William F. DeGrado
PNAS 2016 113 (45 ) pp:E6955-E6964
Publication Date(Web):2016-11-08
DOI:10.1073/pnas.1615471113
The homotetrameric influenza A M2 channel (AM2) is an acid-activated proton channel responsible for the acidification of the
influenza virus interior, an important step in the viral lifecycle. Four histidine residues (His37) in the center of the channel
act as a pH sensor and proton selectivity filter. Despite intense study, the pH-dependent activation mechanism of the AM2
channel has to date not been completely understood at a molecular level. Herein we have used multiscale computer simulations
to characterize (with explicit proton transport free energy profiles and their associated calculated conductances) the activation
mechanism of AM2. All proton transfer steps involved in proton diffusion through the channel, including the protonation/deprotonation
of His37, are explicitly considered using classical, quantum, and reactive molecular dynamics methods. The asymmetry of the
proton transport free energy profile under high-pH conditions qualitatively explains the rectification behavior of AM2 (i.e.,
why the inward proton flux is allowed when the pH is low in viral exterior and high in viral interior, but outward proton
flux is prohibited when the pH gradient is reversed). Also, in agreement with electrophysiological results, our simulations
indicate that the C-terminal amphipathic helix does not significantly change the proton conduction mechanism in the AM2 transmembrane
domain; the four transmembrane helices flanking the channel lumen alone seem to determine the proton conduction mechanism.
Co-reporter:John Savage
The Journal of Physical Chemistry C 2016 Volume 120(Issue 6) pp:3176-3186
Publication Date(Web):January 26, 2016
DOI:10.1021/acs.jpcc.5b11168
Understanding the role of morphology of perfluorosulfonic acid (PFSA) membranes in defining their proton transport behavior is crucial for the development of the next generation of proton exchange membrane fuel cells. In this study, we build large-scale simulation models of three of the most realistic PFSAs morphologies proposed from the results of SAXS experiments and then examine the cation solvation and transport properties of these models. Upon equilibration with molecular dynamics, we find that the bundle morphology immediately flattens into ribbons, in agreement with the locally flat model. The lamellar model, the extreme version of the locally flat model, shows much too fast dynamical properties and too low density. The random model shows acceptable agreement with experiment; however, the bundle model shows the best overall agreement. The addition of sodium as a co-ion to the system makes the water less structured, and while this does not affect water transport it does slows all ionic transport.
Co-reporter:Srabani Taraphder, C. Mark Maupin, Jessica M. J. Swanson, and Gregory A. Voth
The Journal of Physical Chemistry B 2016 Volume 120(Issue 33) pp:8389-8404
Publication Date(Web):April 9, 2016
DOI:10.1021/acs.jpcb.6b02166
The role of protein dynamics in enzyme catalysis is one of the most highly debated topics in enzymology. The main controversy centers around what may be defined as functionally significant conformational fluctuations and how, if at all, these fluctuations couple to enzyme catalyzed events. To shed light on this debate, the conformational dynamics along the transition path surmounting the highest free energy barrier have been herein investigated for the rate limiting proton transport event in human carbonic anhydrase (HCA) II. Special attention has been placed on whether the motion of an excess proton is correlated with fluctuations in the surrounding protein and solvent matrix, which may be rare on the picosecond and subpicosecond time scales of molecular motions. It is found that several active site residues, which do not directly participate in the proton transport event, have a significant impact on the dynamics of the excess proton. These secondary participants are shown to strongly influence the active site environment, resulting in the creation of water clusters that are conducive to fast, moderately slow, or slow proton transport events. The identification and characterization of these secondary participants illuminates the role of protein dynamics in the catalytic efficiency of HCA II.
Co-reporter:Rajib Biswas, Ying-Lung Steve Tse, Andrei Tokmakoff, and Gregory A. Voth
The Journal of Physical Chemistry B 2016 Volume 120(Issue 8) pp:1793-1804
Publication Date(Web):November 17, 2015
DOI:10.1021/acs.jpcb.5b09466
Results from condensed phase ab initio molecular dynamics (AIMD) simulations suggest a proton transfer reaction is facilitated by “presolvation” in which the hydronium is transiently solvated by four water molecules, similar to the typical solvation structure of water, by accepting a weak hydrogen bond from the fourth water molecule. A new version 3.2 multistate empirical valence bond (MS-EVB 3.2) model for the hydrated excess proton incorporating this presolvation behavior is therefore developed. The classical MS-EVB simulations show similar structural properties as those of the previous model but with significantly improved diffusive behavior. The inclusion of nuclear quantum effects in the MS-EVB also provides an even better description of the proton diffusion rate. To quantify the influence of anharmonicity, a second model (aMS-EVB 3.2) is developed using the anharmonic aSPC/Fw water model, which provides similar structural properties but improved spectroscopic responses at high frequencies.
Co-reporter:Glen M. Hocky, Joseph L. Baker, Michael J. Bradley, Anton V. Sinitskiy, Enrique M. De La Cruz, and Gregory A. Voth
The Journal of Physical Chemistry B 2016 Volume 120(Issue 20) pp:4558-4567
Publication Date(Web):May 4, 2016
DOI:10.1021/acs.jpcb.6b02741
Ions regulate the assembly and mechanical properties of actin filaments. Recent work using structural bioinformatics and site-specific mutagenesis favors the existence of two discrete and specific divalent cation binding sites on actin filaments, positioned in the long axis between actin subunits. Cation binding at one site drives polymerization, while the other modulates filament stiffness and plays a role in filament severing by the regulatory protein, cofilin. Existing structural methods have not been able to resolve filament-associated cations, and so in this work we turn to molecular dynamics simulations to suggest a candidate binding pocket geometry for each site and to elucidate the mechanism by which occupancy of the “stiffness site” affects filament mechanical properties. Incorporating a magnesium ion in the “polymerization site” does not seem to require any large-scale change to an actin subunit’s conformation. Binding of a magnesium ion in the “stiffness site” adheres the actin DNase-binding loop (D-loop) to its long-axis neighbor, which increases the filament torsional stiffness and bending persistence length. Our analysis shows that bound D-loops occupy a smaller region of accessible conformational space. Cation occupancy buries key conserved residues of the D-loop, restricting accessibility to regulatory proteins and enzymes that target these amino acids.
Co-reporter:Ying-Lung Steve Tse; Chen Chen; Gerrick E. Lindberg; Revati Kumar
Journal of the American Chemical Society 2015 Volume 137(Issue 39) pp:12610-12616
Publication Date(Web):September 14, 2015
DOI:10.1021/jacs.5b07232
Significant effort has been undertaken to better understand the molecular details governing the propensity of ions for the air–water interface. Facilitated by computationally efficient reactive molecular dynamics simulations, new and statistically conclusive molecular-scale results on the affinity of the hydrated excess proton and hydroxide anion for the air–water interface are presented. These simulations capture the dynamic bond breaking and formation processes (charge defect delocalization) that are important for correctly describing the solvation and transport of these complex species. The excess proton is found to be attracted to the interface, which is correlated with a favorable enthalpic contribution and consistent with reducing the disruption in the hydrogen bond network caused by the ion complex. However, a recent refinement of the underlying reactive potential energy function for the hydrated excess proton shows the interfacial attraction to be weaker, albeit nonzero, a result that is consistent with the experimental surface tension measurements. The influence of a weak hydrogen bond donated from water to the protonated oxygen, recently found to play an important role in excess hydrated proton transport in bulk water, is seen to also be important for this study. In contrast, the hydroxide ion is found to be repelled from the air–water interface. This repulsion is characterized by a reduction of the energetically favorable ion–water interactions, which creates an enthalpic penalty as the ion approaches the interface. Finally, we find that the fluctuation in the coordination number around water sheds new light on the observed entropic trends for both ions.
Co-reporter:Chen Chen; Ying-Lung Steve Tse; Gerrick E. Lindberg; Chris Knight
Journal of the American Chemical Society 2015 Volume 138(Issue 3) pp:991-1000
Publication Date(Web):December 30, 2015
DOI:10.1021/jacs.5b11951
Understanding hydroxide solvation and transport in anion exchange membranes (AEMs) can provide important insight into the design principles of these new membranes. To accurately model hydroxide solvation and transport, we developed a new multiscale reactive molecular dynamics model for hydroxide in aqueous solution, which was then subsequently modified for an AEM material. With this model, we investigated the hydroxide solvation structure and transport mechanism in the membrane. We found that a relatively even separation of the rigid side chains produces a continuous overlapping region for hydroxide transport that is made up of the first hydration shell of the tethered cationic groups. Our results show that hydroxide has a significant preference for this overlapping region, transporting through it and between the AEM side chains with substantial contributions from both vehicular (standard diffusion) and Grotthuss (proton hopping) mechanisms. Comparison of the AEM with common proton exchange membranes (PEMs) showed that the excess charge is less delocalized in the AEM than the PEMs, which is correlated with a higher free energy barrier for proton transfer reactions. The vehicular mechanism also contributes considerably more than the Grotthuss mechanism for hydroxide transport in the AEM, while our previous studies of PEM systems showed a larger contribution from the Grotthuss mechanism than the vehicular mechanism for proton transport. The activation energy barrier for hydroxide diffusion in the AEM is greater than that for proton diffusion in PEMs, implying a more significant enhancement of ion transport in the AEM at elevated temperatures.
Co-reporter:James F. Dama, Glen M. Hocky, Rui Sun, and Gregory A. Voth
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 12) pp:5638-5650
Publication Date(Web):November 3, 2015
DOI:10.1021/acs.jctc.5b00907
Metadynamics is an enhanced sampling method designed to flatten free energy surfaces uniformly. However, the highest-energy regions are often irrelevant to study and dangerous to explore because systems often change irreversibly in unforeseen ways in response to driving forces in these regions, spoiling the sampling. Introducing an on-the-fly domain restriction allows metadynamics to flatten only up to a specified energy level and no further, improving efficiency and safety while decreasing the pressure on practitioners to design collective variables that are robust to otherwise irrelevant high energy driving. This paper describes a new method that achieves this using sequential on-the-fly estimation of energy wells and redefinition of the metadynamics hill shape, termed metabasin metadynamics. The energy level may be defined a priori or relative to unknown barrier energies estimated on-the-fly. Altering only the hill ensures that the method is compatible with many other advances in metadynamics methodology. The hill shape has a natural interpretation in terms of multiscale dynamics, and the computational overhead in simulation is minimal when studying systems of any reasonable size, for instance proteins or other macromolecules. Three example applications show that the formula is accurate and robust to complex dynamics, making metadynamics significantly more forgiving with respect to CV quality and thus more feasible to apply to the most challenging biomolecular systems.
Co-reporter:Jacob W. Wagner, James F. Dama, and Gregory A. Voth
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 8) pp:3547-3560
Publication Date(Web):June 25, 2015
DOI:10.1021/acs.jctc.5b00180
The sensitivity of a coarse-grained (CG) force field to changes in the underlying fine-grained (FG) model from which it was derived provides modeling insight for improving transferability across interaction parameters, transferability across temperature, and the calculation of thermodynamic derivatives. Methods in the literature, such as multi-trajectory finite differences and reweighted finite differences, are either too computationally demanding to calculate within acceptable noise tolerances or are too biased for practical accuracy. This work presents a new reweighting-free, single-simulation formula that allows for practical, high signal-to-noise calculations of CG model sensitivity with respect to FG model interaction parameters and thermodynamic state points. This formula, the self-consistent basis (SCB) single point formula, determines the many-body sensitivity in a single step by approximating the derivative of the many-body potential projected onto the same set of trial functions as the sensitivity. A related diagnostic formula also derived in this paper is the self-consistent iterative (SCI) single point formula, which is useful for identifying the importance of many-body sources of error and verifying CG representability of observables. The SCI formula determines the many-body sensitivity iteratively via a series of partially self-consistent, variational approximations to the complete many-body sensitivity. The new, computationally efficient SCB formula shows substantially less noise than previous methods when applied to single site methanol and solvent-free sodium chloride CG models, though bias can remain a problem. It represents a novel method for calculating alchemical transferability across interaction parameters at low computational cost and with high fidelity, and the results point to new understanding of the current limits of CG model transferability.
Co-reporter:Andrew D. White, James F. Dama, and Gregory A. Voth
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 6) pp:2451-2460
Publication Date(Web):April 30, 2015
DOI:10.1021/acs.jctc.5b00178
Creating models that are consistent with experimental data is essential in molecular modeling. This is often done by iteratively tuning the molecular force field of a simulation to match experimental data. An alternative method is to bias a simulation, leading to a hybrid model composed of the original force field and biasing terms. We previously introduced such a method called experiment directed simulation (EDS). EDS minimally biases simulations to match average values. In this work, we introduce a new method called experiment directed metadynamics (EDM) that creates minimal biases for matching entire free energy surfaces such as radial distribution functions and phi/psi angle free energies. It is also possible with EDM to create a tunable mixture of the experimental data and free energy of the unbiased ensemble with explicit ratios. EDM can be proven to be convergent, and we also present proof, via a maximum entropy argument, that the final bias is minimal and unique. Examples of its use are given in the construction of ensembles that follow a desired free energy. The example systems studied include a Lennard-Jones fluid made to match a radial distribution function, an atomistic model augmented with bioinformatics data, and a three-component electrolyte solution where ab initio simulation data is used to improve a classical empirical model.
Co-reporter:Zhen Cao
The Journal of Physical Chemistry C 2015 Volume 119(Issue 26) pp:14675-14682
Publication Date(Web):March 30, 2015
DOI:10.1021/jp5129244
Water-mediated hydrated proton solvation and diffusion at two types of platinum–water interfaces—namely, the Pt(111) and the Pt(100) surfaces—is investigated using reactive molecular dynamics simulations. The adsorbed water molecules on these platinum surfaces create different hydrogen-bonding networks, resulting in different proton solvation and transport behavior. Free energy calculations show that the excess proton can be stably adsorbed on the Pt(111) surface, while on the Pt(100) surface it prefers to stay at the interface between the hydrophobic layer of adsorbed water and the bulk. The hydrated excess proton can be viewed as a charge defect in the adsorbed water layer, where it diffuses with a low rate due to the slow reorientational dynamics of the adsorbed water molecules. However, the proton can sample a larger surface area by hopping between the adsorbed layer and the bulk at the Pt(111) surface.
Co-reporter:Shule Liu
The Journal of Physical Chemistry C 2015 Volume 119(Issue 4) pp:1753-1762
Publication Date(Web):January 12, 2015
DOI:10.1021/jp511830f
The influence of morphology on proton transport in proton exchange membranes (PEM) is studied at the mesoscale using smoothed particle hydrodynamics (SPH), a mesh-free particle method for solving continuity equations. By solving the Nernst–Planck equation for proton transport in lamellar, cylinder, and cluster morphologies, we find that the proton conductivity for cluster morphology is much lower than lamellar and cylinder morphology at all hydration levels. This suggests the porosity and tortuosity in PEM morphology can reduce proton transport significantly at the mesoscale. We also investigated the effect of including a position-dependent diffusion constant (PDDC) tied to the local morphology, which is usually ignored in studies of proton transport in confinement. We calculated the PDDC in lamellar PEM using both quantitative and phenomenological approaches. SPH calculations show that conductivities for PEM systems with a PDDC can vary compared with systems with uniform diffusion constant. Therefore, it is potentially important to take into account the inhomogeneity of transport coefficients when studying proton transport in anisotropic systems.
Co-reporter:Zhen Cao, Yuxing Peng, and Gregory A. Voth
The Journal of Physical Chemistry B 2015 Volume 119(Issue 24) pp:7516-7521
Publication Date(Web):January 5, 2015
DOI:10.1021/jp511260v
The influence of the Coulomb transport (CT) effect on ion transport in electrolytes is evaluated through two prototypical systems: the [IrCl6]2–/3– redox couple in aqueous solution and Mg2+ (with TFSI– counterions) in acetonitrile solution. In the first system, transport of the [IrCl6]2–/3– anions is inhibited, and ions are trapped near the positive electrode due to the unscreened electric field. With larger applied voltage, the inhibition is stronger. This effect can be weakened, however, by adding supporting charges, which screen the electric field of the electrodes. In the second system, the unscreened electric field provides a means of separating [Mgx(TFSI)y](2x−y)+ clusters in the electrolyte. This effect enhances charge transport by providing a larger number of faster-moving (smaller) ions. Considered together, these systems provide examples of how the CT effect may increase or decrease charge transport and how one might attempt to modulate this effect in order to enhance electrochemical cell conduction.
Co-reporter:Martin McCullagh ; Marissa G. Saunders
Journal of the American Chemical Society 2014 Volume 136(Issue 37) pp:13053-13058
Publication Date(Web):September 2, 2014
DOI:10.1021/ja507169f
Actin performs its myriad cellular functions by the growth and disassembly of its filamentous form. The hydrolysis of ATP in the actin filament has been shown to modulate properties of the filament, thus making it a pivotal regulator of the actin life cycle. Actin has evolved to selectively hydrolyze ATP in the filamentous form, F-actin, with an experimentally observed rate increase over the monomeric form, G-actin, of 4.3 × 104. The cause of this dramatic increase in rate is investigated in this paper using extensive QM/MM simulations of both G- and F-actin. To compute the free energy of hydrolysis in both systems, metadynamics is employed along two collective variables chosen to describe the reaction coordinates of hydrolysis. F-actin is modeled as a monomer with restraints applied to coarse-grained variables enforced to keep it in a filament-like conformation. The simulations reveal a barrier height reduction for ATP hydrolysis in F-actin as compared to G-actin of 8 ± 1 kcal/mol, in good agreement with the experimentally measured barrier height reduction of 7 ± 1 kcal/mol. The barrier height reduction is influenced by an enhanced rotational diffusion of water in F-actin as compared to G-actin and shorter water wires between Asp154 and the nucleophilic water in F-actin, leading to more rapid proton transport.
Co-reporter:Jianing Li ; Brian P. Ziemba ; Joseph J. Falke
Journal of the American Chemical Society 2014 Volume 136(Issue 33) pp:11757-11766
Publication Date(Web):July 30, 2014
DOI:10.1021/ja505369r
Protein kinase C-α (PKCα) has been studied widely as a paradigm for conventional PKCs, with two C1 domains (C1A and C1B) being important for the regulation and function of the kinase. However, it is challenging to explore these domains in membrane-bound environments with either simulations or experiments alone. In this work, we have combined modeling, simulations, and experiments to understand the molecular basis of the PKCα C1A and C1B domain interactions with membranes. Our atomistic simulations of the PKCα C1 domains reveal the dynamic interactions of the proteins with anionic lipids, as well as the conserved hydrogen bonds and the distinct nonpolar contacts formed with lipid activators. Corroborating evidence is obtained from additional simulations and experiments in terms of lipid binding and protein diffusion. Overall, our study, for the first time, explains with atomistic detail how the PKCα C1A and C1B domains interact differently with various lipids. On the molecular level, the information provided by our study helps to shed light on PKCα regulation and activation mechanism. The combined computational/experimental approach demonstrated in this work is anticipated to enable further studies to explore the roles of C1 domains in many signaling proteins and to better understand their molecular mechanisms in normal cellular function and disease development.
Co-reporter:Yuxing Peng, Zhen Cao, Ruhong Zhou, and Gregory A. Voth
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 9) pp:3634-3640
Publication Date(Web):July 25, 2014
DOI:10.1021/ct500447r
An enhanced conformational space sampling method is developed that utilizes replica exchange molecular dynamics between a set of imaginary time Feynman path integral replicas, each having an increasing degree of contraction (or coarse-graining) of the quasi-particle or “polymer beads” in the evaluation of the isomorphic ring-polymer potential energy terms. However, there is no contraction of beads in the effectively harmonic kinetic energy terms. The final replica in this procedure is the fully contracted one in which the potential energy is evaluated only at the centroid of the beads—and hence it is the classical distribution in the centroid variable—while the initial replica has the full degree (or even a heightened degree, if desired) of quantum delocalization and tunneling in the physical potential by the polymer necklace beads. The exchange between the different ring-polymer ensembles is governed by the Metropolis criteria to guarantee detailed balance. The method is applied successfully to several model systems, ranging from one-dimensional prototype rough energy landscape models having analytical solutions to the more realistic alanine dipeptide. A detailed comparison with the classical temperature-based replica exchange method shows an improved efficiency of this new method in the classical conformational space sampling due to coupling with the fictitious path integral (quantum) replicas.
Co-reporter:Anand Srivastava and Gregory A. Voth
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 10) pp:4730-4744
Publication Date(Web):September 10, 2014
DOI:10.1021/ct500474a
We present a methodology to develop coarse-grained lipid models such that electrostatic interactions between the coarse-grained sites can be derived accurately from an all-atom molecular dynamics trajectory and expressed as an effective pairwise electrostatic potential with appropriate screening functions. The reference nonbonded forces from the all-atom trajectory are decomposed into separate electrostatic and van der Waals (vdW) forces, based on the multiscale coarse-graining method. The coarse-grained electrostatic potential is assumed to be a general function of unknown variables and the final site–site interactions are obtained variationally, where the residual of the electrostatic forces from the assumed field is minimized. The resulting electrostatic interactions are fitted to screened electrostatics functions, with a special treatment for distance-dependent dielectrics and screened dipole–dipole interactions. The vdW interactions are derived separately. The resulting charged hybrid coarse-graining method is applied to various solvent-free three-site models of anionic lipid systems.
Co-reporter:Aram Davtyan, James F. Dama, Anton V. Sinitskiy, and Gregory A. Voth
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 12) pp:5265-5275
Publication Date(Web):November 18, 2014
DOI:10.1021/ct500834t
The increasing interest in the modeling of complex macromolecular systems in recent years has spurred the development of numerous coarse-graining (CG) techniques. However, many of the CG models are constructed assuming that all details beneath the resolution of CG degrees of freedom are fast and average out, which sets limits on the resolution of feasible coarse-grainings and on the range of applications of the CG models. Ultra-coarse-graining (UCG) makes it possible to construct models at any desired resolution while accounting for discrete conformational or chemical changes within the CG sites that can modulate the interactions between them. Here, we discuss the UCG methodology and its numerical implementation. We pay particular attention to the numerical mechanism for including state transitions between different conformations within CG sites because this has not been discussed previously. Using a simple example of 1,2-dichloroethane, we demonstrate the ability of the UCG model to reproduce the multiconfigurational behavior of this molecular liquid, even when each molecule is modeled with only one CG site. The methodology can also be applied to other molecular liquids and macromolecular systems with time scale separation between conformational transitions and other intramolecular motions and rotations.
Co-reporter:Andrew D. White and Gregory A. Voth
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 8) pp:3023-3030
Publication Date(Web):June 19, 2014
DOI:10.1021/ct500320c
A primary goal in molecular simulations is to modify the potential energy of a system so that properties of the simulation match experimental data. This is traditionally done through iterative cycles of simulation and reparameterization. An alternative approach is to bias the potential energy so that the system matches experimental data. This can be done while minimally changing the underlying free energy of the molecular simulation. Current minimal biasing methods require replicas, which can lead to unphysical dynamics and introduces new complexity: the choice of replica number and their properties. Here, we describe a new method, called experiment directed simulation that does not require replicas, converges rapidly, can match many data simultaneously, and minimally modifies the potential. The experiment directed simulation method is demonstrated on model systems and a three-component electrolyte simulation. The theory used to derive the method also provides insight into how changing a molecular force-field impacts the expected value of observables in simulation.
Co-reporter:Ruibin Liang, Jessica M. J. Swanson, and Gregory A. Voth
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 1) pp:451-462
Publication Date(Web):December 16, 2013
DOI:10.1021/ct400832r
The self-consistent charge density functional tight binding (SCC-DFTB) method has been increasingly applied to study proton transport (PT) in biological environments. However, recent studies revealing some significant limitations of SCC-DFTB for proton and hydroxide solvation and transport in bulk aqueous systems call into question its accuracy for simulating PT in biological systems. The current work benchmarks the SCC-DFTB/MM method against more accurate DFT/MM by simulating PT in a synthetic leucine–serine channel (LS2), which emulates the structure and function of biomolecular proton channels. It is observed that SCC-DFTB/MM produces overcoordinated and less structured pore water, an overcoordinated excess proton, weak hydrogen bonds around the excess proton charge defect, and qualitatively different PT dynamics. Similar issues are demonstrated for PT in a carbon nanotube, indicating that the inaccuracies found for SCC-DFTB are not due to the point charge based QM/MM electrostatic coupling scheme, but rather to the approximations of the semiempirical method itself. The results presented in this work highlight the limitations of the present form of the SCC-DFTB/MM approach for simulating PT processes in biological protein or channel-like environments, while providing benchmark results that may lead to an improvement of the underlying method.
Co-reporter:John. M. A. Grime and Gregory A. Voth
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 1) pp:423-431
Publication Date(Web):November 5, 2013
DOI:10.1021/ct400727q
The use of coarse-grained (CG) models can significantly increase the time and length scales accessible to computational molecular dynamics (MD) simulations. To address very large-scale phenomena, however, requires a careful consideration of memory requirements and parallel MD load balancing in order to make efficient use of current supercomputers. In this work, a CG-MD code is introduced which is specifically designed for very large, highly parallel simulations of systems with markedly non-uniform particle distributions, such as those found in highly CG models having an implicit solvent. The CG-MD code uses an unorthodox combination of sparse data representations with a Hilbert space-filling curve (SFC) to provide dynamic topological descriptions, reduced memory overhead, and advanced load-balancing characteristics. The results of representative large-scale simulations indicate that our approach can offer significant advantages over conventional MD techniques, and should enable new classes of CG-MD systems to be investigated.
Co-reporter:Ruibin Liang;Hui Li;Jessica M. J. Swanson
PNAS 2014 Volume 111 (Issue 26 ) pp:9396-9401
Publication Date(Web):2014-07-01
DOI:10.1073/pnas.1401997111
The influenza A virus M2 channel (AM2) is crucial in the viral life cycle. Despite many previous experimental and computational
studies, the mechanism of the activating process in which proton permeation acidifies the virion to release the viral RNA
and core proteins is not well understood. Herein the AM2 proton permeation process has been systematically characterized using
multiscale computer simulations, including quantum, classical, and reactive molecular dynamics methods. We report, to our
knowledge, the first complete free-energy profiles for proton transport through the entire AM2 transmembrane domain at various
pH values, including explicit treatment of excess proton charge delocalization and shuttling through the His37 tetrad. The
free-energy profiles reveal that the excess proton must overcome a large free-energy barrier to diffuse to the His37 tetrad,
where it is stabilized in a deep minimum reflecting the delocalization of the excess charge among the histidines and the cost
of shuttling the proton past them. At lower pH values the His37 tetrad has a larger total charge that increases the channel
width, hydration, and solvent dynamics, in agreement with recent 2D-IR spectroscopic studies. The proton transport barrier
becomes smaller, despite the increased charge repulsion, due to backbone expansion and the more dynamic pore water molecules.
The calculated conductances are in quantitative agreement with recent experimental measurements. In addition, the free-energy
profiles and conductances for proton transport in several mutants provide insights for explaining our findings and those of
previous experimental mutagenesis studies.
Co-reporter:Carson J. Bruns;Dr. Jianing Li;Dr. Marco Frasconi;Dr. Severin T. Schneebeli;Dr. Julien Iehl;Dr. Henri-Pierre JacquotdeRouville; Samuel I. Stupp; Gregory A. Voth; J. Fraser Stoddart
Angewandte Chemie International Edition 2014 Volume 53( Issue 7) pp:1953-1958
Publication Date(Web):
DOI:10.1002/anie.201308498
Abstract
Although motor proteins are essential cellular components that carry out biological processes by converting chemical energy into mechanical motion, their functions have been difficult to mimic in artificial synthetic systems. Daisy chains are a class of rotaxanes which have been targeted to serve as artificial molecular machines because their mechanically interlocked architectures enable them to contract and expand linearly, in a manner that is reminiscent of the sarcomeres of muscle tissue. The scope of external stimuli that can be used to control the musclelike motions of daisy chains remains limited, however, because of the narrow range of supramolecular motifs that have been utilized in their templated synthesis. Reported herein is a cyclic daisy chain dimer based on π-associated donor–acceptor interactions, which can be actuated with either thermal or electrochemical stimuli. Molecular dynamics simulations have shown the daisy chain’s mechanism of extension/contraction in the ground state in atomistic detail.
Co-reporter:Carson J. Bruns;Dr. Jianing Li;Dr. Marco Frasconi;Dr. Severin T. Schneebeli;Dr. Julien Iehl;Dr. Henri-Pierre JacquotdeRouville; Samuel I. Stupp; Gregory A. Voth; J. Fraser Stoddart
Angewandte Chemie 2014 Volume 126( Issue 7) pp:1984-1989
Publication Date(Web):
DOI:10.1002/ange.201308498
Abstract
Although motor proteins are essential cellular components that carry out biological processes by converting chemical energy into mechanical motion, their functions have been difficult to mimic in artificial synthetic systems. Daisy chains are a class of rotaxanes which have been targeted to serve as artificial molecular machines because their mechanically interlocked architectures enable them to contract and expand linearly, in a manner that is reminiscent of the sarcomeres of muscle tissue. The scope of external stimuli that can be used to control the musclelike motions of daisy chains remains limited, however, because of the narrow range of supramolecular motifs that have been utilized in their templated synthesis. Reported herein is a cyclic daisy chain dimer based on π-associated donor–acceptor interactions, which can be actuated with either thermal or electrochemical stimuli. Molecular dynamics simulations have shown the daisy chain’s mechanism of extension/contraction in the ground state in atomistic detail.
Co-reporter:John Savage and Gregory A. Voth
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 17) pp:3037-3042
Publication Date(Web):August 20, 2014
DOI:10.1021/jz5014467
Proton transport (PT) in solutions of small amphiphiles in water has previously been shown to be subdiffusive for long times. The present study analyzes simulations of hydrated perfluorosulfonic acid (PFSA) membranes in order to determine whether PT is also subdiffusive in these important amphiphilic systems. We show that PT is indeed subdiffusive for several hundred picoseconds for all hydration levels examined, and the subdiffusive behavior is highly dependent on water concentration. We also investigate the caging of the excess proton using a recently developed technique and show that the excess proton exhibits caging effects up to at least 1 ns in PFSA systems. In order to fully characterize the long-time behavior of PT in PFSAs, these results demonstrate that multiple nanosecond trajectories are needed, well beyond the current capabilities of ab initio molecular dynamics.Keywords: 3M membrane; confined water; proton exchange membrane; proton transport; subdiffusion;
Co-reporter:Ying-Lung Steve Tse, Himanshu N. Sarode, Gerrick E. Lindberg, Thomas A. Witten, Yuan Yang, Andrew M. Herring, and Gregory A. Voth
The Journal of Physical Chemistry C 2014 Volume 118(Issue 2) pp:845-853
Publication Date(Web):December 20, 2013
DOI:10.1021/jp409728a
We have studied anion exchange membrane/polycationic systems with different percentages of fluoride and chloride as counterions by molecular dynamics simulations. We also experimentally measured the self-diffusion constant of fluoride in a diblock copolymer that has the same hydrophilic block and found satisfactory agreement with simulations within a factor of 2. At 300 K, our simulations showed that the self-diffusion constant of fluoride increases by about 70% when fluoride content decreases from 100% to 40% (and 60% Cl), and it increases by about 140% when fluoride content decreases from 100% to 10%. Increasing % Cl also slightly decreases the attraction between fluoride and the cations. We hypothesize that the root cause of the enhancement in fluoride mobility is due to the larger size of the chloride ion, which more readily loses its water solvation shells because of a lower charge/radius squared (surface electric field). This in turn frees up more water for ion transport. We believe this effect is likely more general than just the fluoride/chloride case reported here.
Co-reporter:Himanshu N. Sarode, Gerrick E. Lindberg, Yuan Yang, Lisa E. Felberg, Gregory A. Voth, and Andrew M. Herring
The Journal of Physical Chemistry B 2014 Volume 118(Issue 5) pp:1363-1372
Publication Date(Web):January 20, 2014
DOI:10.1021/jp4085662
This study focuses on understanding the relative effects of ammonium substituent groups (we primarily consider tetramethylammonium, benzyltrimethylammonium, and tetraethylammonium cations) and anion species (OH–, HCO3–, CO32–, Cl–, and F–) on ion transport by combining experimental and computational approaches. We characterize transport experimentally using ionic conductivity and self-diffusion coefficients measured from NMR. These experimental results are interpreted using simulation methods to describe the transport of these cations and anions considering the effects of the counterion. It is particularly noteworthy that we directly probe cation and anion diffusion with pulsed gradient stimulated echo NMR and molecular dynamics simulations, corroborating these methods and providing a direct link between atomic-resolution simulations and macroscale experiments. By pairing diffusion measurements and simulations with residence times, we were able to understand the interplay between short-time and long-time dynamics with ionic conductivity. With experiment, we determined that solutions of benzyltrimethylammonium hydroxide have the highest ionic conductivity (0.26 S/cm at 65 °C), which appears to be due to differences for the ions in long-time diffusion and short-time water caging. We also examined the effect of CO2 on ionic conductivity in ammonium hydroxide solutions. CO2 readily reacts with OH– to form HCO–3 and is found to lower the solution ionic conductivity by almost 50%.
Co-reporter:Zhen Cao, Revati Kumar, Yuxing Peng, and Gregory A. Voth
The Journal of Physical Chemistry B 2014 Volume 118(Issue 28) pp:8090-8098
Publication Date(Web):April 10, 2014
DOI:10.1021/jp501130m
Proton transport through an electrolyte layer between platinum electrodes under a range of applied voltages is explored using reactive molecular dynamics simulation. The proton transport process is decomposed into vehicular and Grotthuss hopping components, and the two mechanisms and their correlation are investigated as a function of applied voltage. At higher applied voltages, the effect of the hopping mechanism is much larger as compared with the vehicular mechanism. As the voltage is increased, the net correlation between the two mechanisms goes from negative to positive, and both the hopping frequency as well as the number of consecutive forward hops increases. This behavior results in a larger total diffusion constant at higher values of the voltage. The behavior of the hydrated excess proton is therefore substantially different under an applied external voltage than in the normal bulk water environment.
Co-reporter:John Savage ; Ying-Lung Steve Tse
The Journal of Physical Chemistry C 2014 Volume 118(Issue 31) pp:17436-17445
Publication Date(Web):July 13, 2014
DOI:10.1021/jp504714d
An understanding of proton transport within perfluorosulfonic acid (PFSA) membranes is crucial to improve the efficiency of proton exchange membrane fuel cells. Using reactive molecular dynamics simulations, we have examined proton transport in two PFSA materials, Hyflon and the 3M membrane, at three different hydration levels. The interaction between the sulfonate group of the polymer side chains and the hydrated protons was found to have only a small influence on the proton transport dynamics. Instead, proton swapping between sulfonate groups is the primary transport mechanism for the proton transport within the pore. The larger water clusters and more flexible side chain of the 3M membrane allows for an enhancement of this swapping mechanism compared to Hyflon. Membranes that can enhance this mechanism may result in greater proton conductivity.
Co-reporter:Jianing Li ; Amanda L. Jonsson ; Thijs Beuming ; John C. Shelley
Journal of the American Chemical Society 2013 Volume 135(Issue 23) pp:8749-8759
Publication Date(Web):May 16, 2013
DOI:10.1021/ja404391q
G-protein-coupled receptors (GPCRs) are membrane proteins with critical functions in cellular signal transduction, representing a primary class of drug targets. Acting by direct binding, many drugs modulate GPCR activity and influence the signaling pathways associated with numerous diseases. However, complete details of ligand-dependent GPCR activation/deactivation are difficult to obtain from experiments. Therefore, it remains unclear how ligands modulate a GPCR’s activity. To elucidate the ligand-dependent activation/deactivation mechanism of the human adenosine A2A receptor (AA2AR), a member of the class A GPCRs, we performed large-scale unbiased molecular dynamics and metadynamics simulations of the receptor embedded in a membrane. At the atomic level, we have observed distinct structural states that resemble the active and inactive states. In particular, we noted key structural elements changing in a highly concerted fashion during the conformational transitions, including six conformational states of a tryptophan (Trp2466.48). Our findings agree with a previously proposed view that, during activation, this tryptophan residue undergoes a rotameric transition that may be coupled to a series of coherent conformational changes, resulting in the opening of the G-protein binding site. Further, metadynamics simulations provide quantitative evidence for this mechanism, suggesting how ligand binding shifts the equilibrium between the active and inactive states. Our analysis also proposes that a few specific residues are associated with agonism/antagonism, affinity, and selectivity, and suggests that the ligand-binding pocket can be thought of as having three distinct regions, providing dynamic features for structure-based design. Additional simulations with AA2AR bound to a novel ligand are consistent with our proposed mechanism. Generally, our study provides insights into the ligand-dependent AA2AR activation/deactivation in addition to what has been found in crystal structures. These results should aid in the discovery of more effective and selective GPCR ligands.
Co-reporter:James F. Dama, Anton V. Sinitskiy, Martin McCullagh, Jonathan Weare, Benoît Roux, Aaron R. Dinner, and Gregory A. Voth
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 5) pp:2466-2480
Publication Date(Web):March 21, 2013
DOI:10.1021/ct4000444
Coarse-grained (CG) models provide a computationally efficient means to study biomolecular and other soft matter processes involving large numbers of atoms correlated over distance scales of many covalent bond lengths and long time scales. Variational methods based on information from simulations of finer-grained (e.g., all-atom) models, for example the multiscale coarse-graining (MS-CG) and relative entropy minimization methods, provide attractive tools for the systematic development of CG models. However, these methods have important drawbacks when used in the “ultra-coarse-grained” (UCG) regime, e.g., at a resolution level coarser or much coarser than one amino acid residue per effective CG particle in proteins. This is due to the possible existence of multiple metastable states “within” the CG sites for a given UCG model configuration. In this work, systematic variational UCG methods are presented that are specifically designed to CG entire protein domains and subdomains into single effective CG particles. This is accomplished by augmenting existing effective particle CG schemes to allow for discrete state transitions and configuration-dependent resolution. Additionally, certain conclusions of this work connect back to single-state force matching and open up new avenues for method development in that area. These results provide a formal statistical mechanical basis for UCG methods related to force matching and relative entropy CG methods and suggest practical algorithms for constructing optimal approximate UCG models from fine-grained simulation data.
Co-reporter:Zhen Cao, James F. Dama, Lanyuan Lu, and Gregory A. Voth
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 1) pp:172-178
Publication Date(Web):November 14, 2012
DOI:10.1021/ct3007277
Solvent free models for aqueous ionic solutions are derived using the multiscale coarse-graining (MS-CG) method to obtain many-body potentials of mean force and generalized Langevin equations to propagate the model in time. The resulting models are compared to other implicit solvent models for aqueous NaCl in terms of both sampling efficiency and accuracy. First, the equilibrium structural properties of the models are compared, and then the temperature dependence of the interion potentials of mean force are determined to obtain the pairwise entropy associated with the effective ionic interactions. After validating the equilibrium behavior of the new models, the dynamical properties are investigated using generalized Langevin equation dynamics simulations. The dynamical properties can be put into better agreement with the original atomistic models by introducing an exponential memory kernel to account for the strong coupling between the ions and their water solvation shells.
Co-reporter:Anand Srivastava and Gregory A. Voth
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 1) pp:750-765
Publication Date(Web):November 9, 2012
DOI:10.1021/ct300751h
We present a systematic methodology to develop highly coarse-grained (CG) lipid models for large scale biomembrane simulations, in which we derive CG interactions using a powerful combination of the multiscale coarse-graining (MS-CG) method, and an analytical form of the CG potential to model interactions at short-range. The resulting hybrid coarse-graining (HCG) methodology is used to develop a three-site solvent-free model for 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), and a 1:1 mixture of 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (DOPS) and DOPC. In addition, we developed a four-site model of DOPC, demonstrating the capability of the HCG methodology in designing model lipid systems of a desired resolution. We carried out microsecond-scale molecular dynamics (MD) simulations of large vesicles, highlighting the ability of the model to study systems at mesoscopic length and time scales. The models of DLPC, DOPC, and DOPC/DOPS have elastic properties consistent with experiment and structural properties such as the radial distribution functions (RDF), bond and angle distributions, and the z-density distributions that compare well with reference all-atom systems.
Co-reporter:Adrian W. Lange and Gregory A. Voth
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 9) pp:4018-4025
Publication Date(Web):August 14, 2013
DOI:10.1021/ct400516x
We introduce a multistate framework for Fragment Molecular Orbital (FMO) quantum mechanical calculations and implement it in the context of protonated water clusters. The purpose of the framework is to address issues of nonuniqueness and dynamic fragmentation in FMO as well as other related fragment methods. We demonstrate that our new approach, Fragment Molecular Orbital Multistate Reactive Molecular Dynamics (FMO-MS-RMD), can improve energetic accuracy and yield stable molecular dynamics for small protonated water clusters undergoing proton transfer reactions.
Co-reporter:Anton V. Sinitskiy, Gregory A. Voth
Chemical Physics 2013 Volume 422() pp:165-174
Publication Date(Web):30 August 2013
DOI:10.1016/j.chemphys.2013.01.024
Abstract
To simulate molecular processes on biologically relevant length- and timescales, coarse-grained (CG) models of biomolecular systems with tens to even hundreds of residues per CG site are required. One possible way to build such models is explored in this article: an elastic network model (ENM) is employed to define the CG variables. Free energy surfaces are approximated by Taylor series, with the coefficients found by force-matching. CG potentials are shown to undergo renormalization due to roughness of the energy landscape and smoothing of it under coarse-graining. In the case study of hen egg-white lysozyme, the entropy factor is shown to be of critical importance for maintaining the native structure, and a relationship between the proposed ENM-mode-based CG models and traditional CG-bead-based models is discussed. The proposed approach uncovers the renormalizable character of CG models and offers new opportunities for automated and computationally efficient studies of complex free energy surfaces.
Co-reporter:Tae Hoon Choi, Ruibin Liang, C. Mark Maupin, and Gregory A. Voth
The Journal of Physical Chemistry B 2013 Volume 117(Issue 17) pp:5165-5179
Publication Date(Web):April 8, 2013
DOI:10.1021/jp400953a
The self-consistent charge density functional tight binding (SCC-DFTB) method has been applied to hydroxide water clusters and a hydroxide ion in bulk water. To determine the impact of various implementations of SCC-DFTB on the energetics and dynamics of a hydroxide ion in gas phase and condensed phase, the DFTB2, DFTB2-γh, DFTB2-γh+gaus, DFTB3-diag, DFTB3-diag+gaus, DFTB3-Full+gaus, and DFTB3-3OB implementations have been tested. Energetic stabilities for small hydroxide clusters, OH–(H2O)n, where n = 4–7, are inconsistent with the results calculated with the B3LYP and second order Møller–Plesset (MP2) levels of ab initio theory. The condensed phase simulations, OH–(H2O)127, using the DFTB2, DFTB2-γh, DFTB2-γh+gaus, DFTB3-diag, DFTB3-diag+gaus, DFTB3-Full+gaus and DFTB3-3OB methods are compared to Car–Parrinello molecular dynamics (CPMD) simulations using the BLYP functional. The SCC-DFTB method including a modified O–H repulsive potential and the third order correction (DFTB3-diag/Full+gaus) is shown to poorly reproduce the CPMD computational results, while the DFTB2 and DFTB2-γh method somewhat more closely describe the structural and dynamical nature of the hydroxide ion in condensed phase. The DFTB3-3OB outperforms the MIO parameter set but is no more accurate than DFTB2. It is also shown that the overcoordinated water molecules lead to an incorrect bulk water density and result in unphysical water void formation. The results presented in this paper point to serious drawbacks for various DFTB extensions and corrections for a hydroxide ion in aqueous environments.
Co-reporter:Martin McCullagh and Gregory A. Voth
The Journal of Physical Chemistry B 2013 Volume 117(Issue 15) pp:4062-4071
Publication Date(Web):March 14, 2013
DOI:10.1021/jp402441s
Hydrogenase enzymes are natural biocatalysts that might be harnessed to reduce the cost of hydrogen gas production. [FeFe]-hydrogenases are the most effective of three such enzymes at catalyzing H+ reduction. In this study, we develop and apply a novel combination of all-atom molecular dynamics and coarse-grained (CG) analysis to characterize two important steps of the catalytic cycle of [FeFe]-hydrogenase. The first is the electron transport through FeS clusters to the active site. We use a Marcus formulation to compute the free energy and the reorganization energy of three electron transport steps and decompose these values into contributions from the CG protein sites and the solvent. The three-step transport process is found to be downhill with relative free energies of −11.7 for the first step, −14.8 for the second step, and −17.1 kcal/mol for the third step. The electron-transport process is also found to activate a water channel suggesting a coupled mechanism for proton and electron transport to the active site. The channel opening is orchestrated by three CG sites located in the active-site domain of the protein with one of the sites also contributing a strong attractive electrostatic potential (ESP) to help shuttle protons to the active site. Overall, our CG analysis points to a concerted mechanism of electron and proton delivery to the active site in these proteins thus providing important insight for the development of biomimetic devices.
Co-reporter:Ying-Lung Steve Tse ; Andrew M. Herring ; Kwiseon Kim
The Journal of Physical Chemistry C 2013 Volume 117(Issue 16) pp:8079-8091
Publication Date(Web):April 4, 2013
DOI:10.1021/jp400693g
Proton transfer and local structures in 3M (EW 825) and Nafion (EW 890) membranes are investigated in this study by both standard nonreactive molecular dynamics and the self-consistent iterative multistate empirical valence bond method, which is capable of simulating multiple reactive protons and accounting for the Grotthuss mechanism of proton transport. The Nafion and 3M systems have the same backbone, so we can isolate and compare the effect of the different side chains by calculating the radial distribution functions (RDFs), self-diffusion constants, and other properties for three hydration levels at 5, 9, and 14 at 300 and 353 K. The conformations of the 3M and Nafion side chains are also compared. We found that even though many results are similar for both F3C and SPC/Fw water models, certain trends such as the sulfonate clustering can depend on the water model selected. The relationship between the different RDFs for the sulfonate, water, and hydronium is discussed. The self-diffusion constants of water for both membranes are found to be close with respect to each water model selected, even though the experimental values for 3M at 300 K are higher. The calculated self-diffusion constants of the excess protons are found to be higher for 3M than Nafion for hydration levels 9 and 14 at 300 K but statistically the same at 353 K.
Co-reporter:Ryan Jorn, Revati Kumar, Daniel P. Abraham, and Gregory A. Voth
The Journal of Physical Chemistry C 2013 Volume 117(Issue 8) pp:3747-3761
Publication Date(Web):January 25, 2013
DOI:10.1021/jp3102282
The solid electrolyte interface (SEI) forms as a result of side reactions between the electrolyte and electrode surfaces in Li-ion batteries and can adversely impact performance by impeding Li-ion transport and diminishing the storage capacity of the battery. To gain a detailed understanding of the impact of the SEI on electrolyte structure, atomistic molecular dynamics simulations of the electrode/electrolyte interface were performed in the presence and absence of the SEI under applied voltages. The composition of the SEI was guided by a wealth of data from experiments and allowed to vary across the simulations. A novel computational approach was implemented that showed significant computational speedup compared to fully polarizable electrode simulations, yet, retained the correct qualitative physics for the electrolyte. A force-matching algorithm was used to construct a new force field for the pure electrolyte, LiPF6 in ethylene carbonate, which was developed from ab initio molecular dynamics simulations. The electrode/electrolyte interface was included using a simple, physically motivated model, which includes the polarization of the conducting graphitic electrode by the electrolyte and the application of an external voltage. Changes in the structure of the electrolyte at the interface as a function of applied voltage, the thickness of the SEI layer, and composition of the SEI provide molecular level insight into the species present at these interfaces and potential clues to the effect of the SEI on transport. It is noted that, with increasing SEI thickness and LiF content, lithium ions are drawn closer to the SEI surface, which implies that these interfaces favor desolvation and promote more rapid lithium transport.
Co-reporter:Mijo Simunovic;Anand Srivastava
PNAS 2013 Volume 110 (Issue 51 ) pp:20396-20401
Publication Date(Web):2013-12-17
DOI:10.1073/pnas.1309819110
Adhesion and insertion of curvature-mediating proteins can induce dramatic structural changes in cell membranes, allowing
them to participate in several key cellular tasks. The way proteins interact to generate curvature remains largely unclear,
especially at early stages of membrane remodeling. Using a coarse-grained model of Bin/amphiphysin/Rvs domain with an N-terminal
helix (N-BAR) interacting with flat membranes and vesicles, we demonstrate that at low protein surface densities, binding
of N-BAR domain proteins to the membrane is followed by a linear aggregation and the formation of meshes on the surface. In
this process, the proteins assemble at the base of emerging membrane buds. Our work shows that beyond a more straightforward
scaffolding mechanism at high bound densities, the interplay of anisotropic interactions and the local stress imposed by the
N-BAR proteins results in deep invaginations and endocytic vesicular bud-like deformations, an order of magnitude larger than
the size of the individual protein. Our results imply that by virtue of this mechanism, cell membranes may achieve rapid local
increases in protein concentration.
Co-reporter:Anuj Chaudhri, Isidro E. Zarraga, Sandeep Yadav, Thomas W. Patapoff, Steven J. Shire, and Gregory A. Voth
The Journal of Physical Chemistry B 2013 Volume 117(Issue 5) pp:1269-1279
Publication Date(Web):January 14, 2013
DOI:10.1021/jp3108396
Coarse-grained computational models of therapeutic monoclonal antibodies and their mutants can be used to understand the effect of domain-level charge–charge electrostatics on the self-association phenomena at high protein concentrations. The coarse-grained models are constructed for two antibodies at different coarse-grained resolutions by using six different concentrations. It is observed that a particular monoclonal antibody (hereafter referred to as MAb1) forms three-dimensional heterogeneous structures with dense regions or clusters compared to a different monoclonal antibody (hereafter referred to as MAb2) that forms homogeneous structures without clusters. The potential of mean force (PMF) and radial distribution functions (RDF) plots for the mutants (hereafter referred to as M1, M5, M7, and M10) show trends consistent with previously reported experimental observation of viscosities. The mutant referred to as M6 shows strongly attractive interactions that are consistent with previously reported negative second virial coefficients (B22) obtained from light-scattering experiments (Yadav et al. Pharm. Res.2011, 28, 1750–1764; Yadav et al. Mol. Pharmaceutics.2012, 9, 791–802). Clustering data on MAb1 reveal a small number of large clusters that are hypothesized to be the reason for the high experimental viscosity. This is in contrast with M6 (that differs from MAb1 in only a few amino acids), where cluster analysis reveals the formation of a large number of smaller clusters that is hypothesized to be the reason for the observed lower viscosity. The coarse-grained representations are effective in picking up differences based on local charge distributions of domains to make predictions on the self-association characteristics of these protein solutions.
Co-reporter:Jianqing Xu, Takefumi Yamashita, Noam Agmon, and Gregory A. Voth
The Journal of Physical Chemistry B 2013 Volume 117(Issue 49) pp:15426-15435
Publication Date(Web):July 3, 2013
DOI:10.1021/jp4051726
Recent experiments reported that proton mobility in tetramethylurea (TMU) solutions is much slower than in urea solutions of the same molarity, and this (as well as the significantly retarded water reorientation) was ascribed to hydrophopic effects. In order to further explore the mechanism of proton transport in these solutions, reactive molecular dynamics simulations using a multistate empirical valence bond model were conducted. The simulations showed that the hydrophobic effect of the TMU methyl groups is weaker than believed. Rather, water concentration is the dominant factor determining proton diffusion. This contrasts with water reorientation and self-diffusion in these samples, which are mutually correlated and depend on the identity of the solute. Interestingly, we find that the mean squared displacements (MSDs) of both water and proton grow nonlinearly in time up to at least 1 ns (“transient subdiffusion”). Subdiffusion is more pronounced for the proton, with an exponent as low as 0.85 that depends, again, on water concentration. Hence, the experimentally relevant long-time diffusivity is observably smaller than what is usually deduced from short simulation runs. It exhibits, for both water and proton, a universal dependence on the power-law exponent.
Co-reporter:Chris Knight and Gregory A. Voth
Accounts of Chemical Research 2012 Volume 45(Issue 1) pp:101
Publication Date(Web):August 22, 2011
DOI:10.1021/ar200140h
Understanding the hydrated proton is a critically important problem that continues to engage the research efforts of chemists, physicists, and biologists because of its involvement in a wide array of phenomena. Only recently have several unique properties of the hydrated proton been unraveled through computer simulations. One such process is the detailed molecular mechanism by which protons hop between neighboring water molecules, thus giving rise to the anomalously high diffusion of protons relative to other simple cations. Termed Grotthuss shuttling, this process occurs over multiple time and length scales, presenting unique challenges for computer modeling and simulation. Because the hydrated proton is in reality a dynamical electronic charge defect that spans multiple water molecules, the simulation methodology must be able to dynamically readjust the chemical bonding topology. This reactive nature of the chemical process is automatically captured with ab initio molecular dynamics (AIMD) simulation methods, where the electronic degrees of freedom are treated explicitly. Unfortunately, these calculations can be prohibitively expensive for more complex proton solvation and transport phenomena in the condensed phase. These AIMD simulations remain extremely valuable, however, in validating empirical models, verifying results, and providing insight into molecular mechanisms.In this Account, we discuss recent progress in understanding the solvation and transport properties of the hydrated excess proton. The advances are based on results obtained from reactive molecular dynamics simulations using the multistate empirical valence bond (MS-EVB) methodology. This approach relies on a dynamic linear combination of chemical bond topologies to model charge delocalization and dynamic bonding environments. When parametrized via a variational force-matching algorithm from AIMD trajectories, the MS-EVB method can be viewed as a multiscale bridging of ab initio simulation results to a simpler and more efficient representation. The process allows sampling of longer time and length scales, which would normally be too computationally expensive with AIMD alone.With the MS-EVB methodology, the statistically important components of the excess proton solvation and hopping mechanisms in liquid water have been identified. The most likely solvation structure for the hydrated proton is a distorted Eigen-type complex (H9O4+). In this state, the excess proton charge defect rapidly resonates between three possible distorted Eigen-type structures until a successful proton hop occurs. This process, termed the “special-pair dance”, serves as a kind of preparatory phase for the proton hopping while the neighboring water hydrogen-bonding network fluctuates and ultimately rearranges to facilitate a proton hop.The modifications of the solvation structure and transport properties of the excess proton in concentrated acid solutions were further investigated. The Eigen-type solvation structure also possesses both “hydrophilic” and “hydrophobic” sides, which accounts for the affinity of the hydrated proton for the air–water interface. This unusual “amphiphilic” character of the hydrated proton further leads to the metastable formation of contact ion pairs between two hydrated protons. It also engenders a surprisingly constant degree of solubility of hydrophobic species as a function of acid concentration, which contrasts with a markedly variable solubility as a function of salt (such as NaCl or KCl) concentration.
Co-reporter:Ryan Jorn, John Savage, and Gregory A. Voth
Accounts of Chemical Research 2012 Volume 45(Issue 11) pp:2002
Publication Date(Web):May 17, 2012
DOI:10.1021/ar200323q
Concerns over global climate change associated with fossil-fuel consumption continue to drive the development of electrochemical alternatives for energy technology. Proton exchange fuel cells are a particularly promising technology for stationary power generation, mobile electronics, and hybrid engines in automobiles. For these devices to work efficiently, direct electrical contacts between the anode and cathode must be avoided; hence, the separator material must be electronically insulating but highly proton conductive. As a result, researchers have examined a variety of polymer electrolyte materials for use as membranes in these systems.In the optimization of the membrane, researchers are seeking high proton conductivity, low electronic conduction, and mechanical stability with the inclusion of water in the polymer matrix. A considerable number of potential polymer backbone and side chain combinations have been synthesized to meet these requirements, and computational studies can assist in the challenge of designing the next generation of technologically relevant membranes. Such studies can also be integrated in a feedback loop with experiment to improve fuel cell performance. However, to accurately simulate the currently favored class of membranes, perfluorosulfonic acid containing moieties, several difficulties must be addressed including a proper treatment of the proton-hopping mechanism through the membrane and the formation of nanophase-separated water networks.We discuss our recent efforts to address these difficulties using methods that push the limits of computer simulation and expand on previous theoretical developments. We describe recent advances in the multistate empirical valence bond (MS-EVB) method that can probe proton diffusion at the nanometer-length scale and accurately model the so-called Grotthuss shuttling mechanism for proton diffusion in water. Using both classical molecular dynamics and coarse-grained descriptions that replace atomistic representations with collective coordinates, we investigated the proton conductivity of polymer membrane structure as a function of hydration level. Nanometer-sized water channels form torturous pathways that are traversed by the charges during fuel cell operation. Using a combination of coarse-grained membrane structure and novel multiscale methods, we demonstrate emerging approaches to treat proton motion at the mesoscale in these complex materials.
Co-reporter:Takefumi Yamashita, Yuxing Peng, Chris Knight, and Gregory A. Voth
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 12) pp:4863-4875
Publication Date(Web):August 21, 2012
DOI:10.1021/ct3006437
It is a computationally demanding task to explicitly simulate the electronic degrees of freedom in a system to observe the chemical transformations of interest, while at the same time sampling the time and length scales required to converge statistical properties and thus reduce artifacts due to initial conditions, finite-size effects, and limited sampling. One solution that significantly reduces the computational expense consists of molecular models in which effective interactions between particles govern the dynamics of the system. If the interaction potentials in these models are developed to reproduce calculated properties from electronic structure calculations and/or ab initio molecular dynamics simulations, then one can calculate accurate properties at a fraction of the computational cost. Multiconfigurational algorithms model the system as a linear combination of several chemical bonding topologies to simulate chemical reactions, also sometimes referred to as “multistate”. These algorithms typically utilize energy and force calculations already found in popular molecular dynamics software packages, thus facilitating their implementation without significant changes to the structure of the code. However, the evaluation of energies and forces for several bonding topologies per simulation step can lead to poor computational efficiency if redundancy is not efficiently removed, particularly with respect to the calculation of long-ranged Coulombic interactions. This paper presents accurate approximations (effective long-range interaction and resulting hybrid methods) and multiple-program parallelization strategies for the efficient calculation of electrostatic interactions in reactive molecular simulations.
Co-reporter:Isaiah Sumner and Gregory A. Voth
The Journal of Physical Chemistry B 2012 Volume 116(Issue 9) pp:2917-2926
Publication Date(Web):February 7, 2012
DOI:10.1021/jp208512y
Hydrogenases reversibly catalyze the production of molecular hydrogen. Current interest in these enzymes is focused on understanding the catalysis, since this may prove useful for hydrogen-based fuel cell and photosynthetic hydrogen production cell technologies. A key step in the hydrogenase catalytic cycle and the focus of this work is proton transport (PT) to and from the active site. The PT mechanism of the enzyme is studied using reactive molecular dynamics simulations of the full protein and the excess proton transfers via the multistate empirical valence bond (MS-EVB) method. Pathways connecting the bulk and the active site are located that suggest possible participation by several protonatable residues. PT free energy surfaces are calculated to differentiate the pathways.
Co-reporter:Fehmi Bardak;Dr. Dong Xiao;Larry G. Hines Jr.;Dr. Pillhun Son; Richard A. Bartsch; Edward L. Quitevis;Peng Yang; Gregory A. Voth
ChemPhysChem 2012 Volume 13( Issue 7) pp:1687-1700
Publication Date(Web):
DOI:10.1002/cphc.201200026
Abstract
The nanostructural organization and subpicosecond intermolecular dynamics in mixtures of acetonitrile and the ionic liquid (IL) 1-pentyl-3-methylimidazolium bis{(trifluoromethane)sulfonyl}amide ([C5mim][NTf2]) are studied as a function of concentration using molecular dynamics (MD) simulations and optical heterodyne-detected Raman-induced Kerr effect spectroscopy. The MD simulations show the IL to be nanostructurally organized into an ionic network and nonpolar domains, with CH3CN molecules localized in the interfacial region between the ionic network and nonpolar domains, as found previously by other researchers. The MD simulations indicate strong interactions between CH3CN and the hydrogen atoms on the imidazolium ring of the cation. The low-frequency (0–200 cm−1) intermolecular part of the reduced spectral densities (RSDs) of the mixtures narrows and shifts to lower frequency as the concentration of CH3CN increases. These spectral changes can be partly attributed to the increasing contribution of the low-frequency intermolecular modes of CH3CN to the RSD. At a given composition, the RSD of a mixture is found to be broader and higher in frequency than the corresponding ideal RSD given by the volume-fraction-weighted sum of the RSDs of the neat liquids. This difference is rationalized in terms of the competition between CH3CN–cation interactions and solute-induced disruption of the ionic networks.
Co-reporter:Shulu Feng, John Savage, and Gregory A. Voth
The Journal of Physical Chemistry C 2012 Volume 116(Issue 36) pp:19104-19116
Publication Date(Web):August 20, 2012
DOI:10.1021/jp304783z
The effects of polymer morphology on proton solvation and transport in hydrated Nafion are investigated by using a novel reactive molecular dynamics approach. Three of the most significant morphological models of Nafion, the lamellar model, the cylinder model, and the cluster-channel model, are studied. The three models exhibit distinct proton transport (PT) patterns, which result in different proton diffusion rates. In both the lamellar and the cylinder models, the interaction between protons and the sulfonate groups is shown to be the key factor in determining PT behavior. For the cluster-channel model, the geometrical shape also plays an important role in influencing the PT behavior. The change in the excess proton solvation structure as a function of the distance between protons and sulfonate groups is also analyzed. It is found that the increase of the water cylinder radius or water layer height leads to the presence of more protons around the sulfonate groups, while, for the cluster model, an increase in the water sphere radius leads to the presence of fewer protons around the sulfonate groups. Furthermore, for the lamellar and cylinder models, the hydrated protons around the sulfonate groups consist of more Zundel-like (H5O2+) structures when the hydration levels decrease, which is also influenced by the different morphological structures of Nafion.
Co-reporter:Fehmi Bardak;Dr. Dong Xiao;Larry G. Hines Jr.;Dr. Pillhun Son; Richard A. Bartsch; Edward L. Quitevis;Peng Yang; Gregory A. Voth
ChemPhysChem 2012 Volume 13( Issue 7) pp:
Publication Date(Web):
DOI:10.1002/cphc.201290029
Co-reporter:Anuj Chaudhri, Isidro E. Zarraga, Tim J. Kamerzell, J. Paul Brandt, Thomas W. Patapoff, Steven J. Shire, and Gregory A. Voth
The Journal of Physical Chemistry B 2012 Volume 116(Issue 28) pp:8045-8057
Publication Date(Web):June 13, 2012
DOI:10.1021/jp301140u
Coarse-grained computational models of two therapeutic monoclonal antibodies are constructed to understand the effect of domain-level charge–charge electrostatics on the self-association phenomena at high protein concentrations. The coarse-grained representations of the individual antibodies are constructed using an elastic network normal-mode analysis. Two different models are constructed for each antibody for a compact Y-shaped and an extended Y-shaped configuration. The resulting simulations of these coarse-grained antibodies that interact through screened electrostatics are done at six different concentrations. It is observed that a particular monoclonal antibody (hereafter referred to as MAb1) forms three-dimensional heterogeneous structures with dense regions or clusters compared to a different monoclonal antibody (hereafter referred to as MAb2) that forms more homogeneous structures (no clusters). These structures, together with the potential mean force (PMF) and radial distribution functions (RDF) between pairs of coarse-grained regions on the MAbs, are qualitatively consistent with the experimental observation that MAb1 has a significantly higher viscosity compared to MAb2, especially at concentrations >50 mg/mL, even though the only difference between the MAbs lies with a few amino acids at the antigen-binding loops (CDRs). It is also observed that the structures in MAb1 are formed due to stronger Fab–Fab interactions in corroboration with experimental observations. Evidence is also shown that Fab–Fc interactions can be equally important in addition to Fab–Fab interactions. The coarse-grained representations are effective in picking up differences based on local charge distributions of domains and make predictions on the self-association characteristics of these protein solutions. This is the first computational study of its kind to show that there are differences in structures formed by two different monoclonal antibodies at high concentrations.
Co-reporter:Anton V. Sinitskiy, Marissa G. Saunders, and Gregory A. Voth
The Journal of Physical Chemistry B 2012 Volume 116(Issue 29) pp:8363-8374
Publication Date(Web):January 25, 2012
DOI:10.1021/jp2108895
The computational study of large biomolecular complexes (molecular machines, cytoskeletal filaments, etc.) is a formidable challenge facing computational biophysics and biology. To achieve biologically relevant length and time scales, coarse-grained (CG) models of such complexes usually must be built and employed. One of the important early stages in this approach is to determine an optimal number of CG sites in different constituents of a complex. This work presents a systematic approach to this problem. First, a universal scaling law is derived and numerically corroborated for the intensity of the intrasite (intradomain) thermal fluctuations as a function of the number of CG sites. Second, this result is used for derivation of the criterion for the optimal number of CG sites in different parts of a large multibiomolecule complex. In the zeroth-order approximation, this approach validates the empirical rule of taking one CG site per fixed number of atoms or residues in each biomolecule, previously widely used for smaller systems (e.g., individual biomolecules). The first-order corrections to this rule are derived and numerically checked by the case studies of the Escherichia coli ribosome and Arp2/3 actin filament junction. In different ribosomal proteins, the optimal number of amino acids per CG site is shown to differ by a factor of 3.5, and an even wider spread may exist in other large biomolecular complexes. Therefore, the method proposed in this paper is valuable for the optimal construction of CG models of such complexes.
Co-reporter:Matt K. Petersen ; Revati Kumar ; Henry S. White
The Journal of Physical Chemistry C 2012 Volume 116(Issue 7) pp:4903-4912
Publication Date(Web):January 12, 2012
DOI:10.1021/jp210252g
A computationally efficient method is presented for the treatment of electrostatic interactions between polarizable metallic electrodes held at a constant potential and separated by an electrolyte. The method combines a fluctuating uniform electrode charge with explicit image charges to account for the polarization of the electrode by the electrolyte, and a constant uniform charge added to the fluctuating uniform electrode charge to account for the constant potential condition. The method is then used to calculate electron transport rates using Marcus theory and these rates are incorporated in a Kinetic Monte Carlo - Molecular Dynamics simulation scheme to efficiently model oxidation/reduction reactions in an electrochemical cell.
Co-reporter:Ryan Jorn
The Journal of Physical Chemistry C 2012 Volume 116(Issue 19) pp:10476-10489
Publication Date(Web):March 14, 2012
DOI:10.1021/jp300040w
Previous efforts to model proton transport through fuel cell membranes have largely focused on disparate length scales: molecular dynamics at the atomistic level and fuel cell stack engineering approaches at the macroscale. A new multiscale approach to bridge these extremes is proposed in this work which combines concepts from coarse-grained (CG) modeling with smoothed particle hydrodynamics (SPH) to capture the qualitative morphology and transport behavior of a proton exchange membrane at the length scale of tens of nanometers. This method allows for connection to atomistic simulations via the inclusion of transport coefficients from molecular dynamics and coarse-grained forces derived for the polymer backbone, side chain, proton, and water interactions. Information pertaining to macroscopic conductivity is obtained by volume averaging based on the flux and chemical potential fields within the membrane. Proton transport is effectively coarse-grained via introduction of a composition variable associated with each interacting site which carries the field information. By combining this technique with local electrostatics and coordinate dependent diffusion constants, the effects of double layer formation within the water pores and the influence of proximity to sulfonate groups on transport is recovered. The combined CG-SPH method is validated and subsequently applied to an equilibrated hydrated Nafion structure with a box length of 40 nm. The resulting conductivities calculated for the material agree very well with trends from experiment and provide insight into the complex interplay of morphology, proton distribution, and diffusion coefficients at a length scale that can be expanded beyond feasible atomistic molecular dynamics simulations to capture the effects of mesoscopic morphology on proton conduction.
Co-reporter:Zhiyong Zhang ; Karissa Y. Sanbonmatsu
Journal of the American Chemical Society 2011 Volume 133(Issue 42) pp:16828-16838
Publication Date(Web):September 12, 2011
DOI:10.1021/ja2028487
The ribosome is a very large complex that consists of many RNA and protein molecules and plays a central role in protein biosynthesis in all organisms. Extensive interactions between different molecules are critical to ribosomal functional dynamics. In this work, intermolecular interactions in the Escherichia coli 70S ribosome are investigated by coarse-grained (CG) analysis. CG models are defined to preserve dynamic domains in RNAs and proteins and to capture functional motions in the ribosome, and then the CG sites are connected by harmonic springs, and spring constants are obtained by matching the computed fluctuations to those of an all-atom molecular dynamics (MD) simulation. Those spring constants indicate how strong the interactions are between the ribosomal components, and they are in good agreement with various experimental data. Nearly all the bridges between the small and large ribosomal subunits are indicated by CG interactions with large spring constants. The head of the small subunit is very mobile because it has minimal CG interactions with the rest of the subunit; however, a large number of small subunit proteins bind to maintain the internal structure of the head. The results show a clear connection between the intermolecular interactions and the structural and functional properties of the ribosome because of the reduced complexity in domain-based CG models. The present approach also provides a useful strategy to map interactions between molecules within large biomolecular complexes since it is not straightforward to investigate these by either atomistic MD simulations or residue-based elastic network models.
Co-reporter:C. Mark Maupin ; Norberto Castillo ; Srabani Taraphder ; Chingkuang Tu ; Robert McKenna ; David N. Silverman
Journal of the American Chemical Society 2011 Volume 133(Issue 16) pp:6223-6234
Publication Date(Web):March 31, 2011
DOI:10.1021/ja1097594
In human carbonic anhydrase II (HCA II), the mutation of position 64 from histidine to alanine (H64A) disrupts the rate limiting proton transfer (PT) event, resulting in a reduction of the catalytic activity of the enzyme as compared to the wild-type. Potential of mean force (PMF) calculations utilizing the multistate empirical valence bond (MS-EVB) methodology for H64A HCA II yields a PT free energy barrier significantly higher than that found in the wild-type enzyme. This high barrier, determined in the absence of exogenous buffer and assuming no additional ionizable residues in the PT pathway, indicates the likelihood of alternate enzyme pathways that utilize either ionizable enzyme residues (self-rescue) and/or exogenous buffers (chemical rescue). It has been shown experimentally that the catalytic activity of H64A HCA II can be chemically rescued to near wild-type levels by the addition of the exogenous buffer 4-methylimidazole (4MI). Crystallographic studies have identified two 4MI binding sites, yet site-specific mutations intended to disrupt 4MI binding have demonstrated these sites to be nonproductive. In the present work, MS-EVB simulations show that binding of 4MI near Thr199 in the H64A HCA II mutant, a binding site determined by NMR spectroscopy, results in a viable chemical rescue pathway. Additional viable rescue pathways are also identified where 4MI acts as a proton transport intermediary from the active site to ionizable residues on the rim of the active site, revealing a probable mode of action for the chemical rescue pathway.
Co-reporter:Yong Zhang and Gregory A. Voth
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 7) pp:2277-2283
Publication Date(Web):June 2, 2011
DOI:10.1021/ct200100e
Free energy calculations are one of the most useful methods for the study of ion transport mechanisms through confined spaces such as protein ion channels. Their reliability depends on a correctly defined reaction coordinate (RC). A straight line is usually not a proper RC for such complicated processes, so in this work a combined metadynamics/umbrella sampling (MTD/US) method is proposed. In the combined method, the ion transport pathway is first identified by the MTD method, and then the free energy profile or potential of mean force (PMF) along the path is calculated using umbrella sampling. This combined method avoids the discontinuity problem often associated with normal umbrella sampling calculations that assume a straight line RC, and it provides a more physically accurate potential of mean force for such processes. The method is demonstrated for the proton transport process through the protein channel of aquaporin-1.
Co-reporter:Edward Lyman, Haosheng Cui and Gregory A. Voth
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 22) pp:10430-10436
Publication Date(Web):18 Apr 2011
DOI:10.1039/C0CP02978E
We present a method for “inverse coarse graining,” rebuilding a higher resolution model from a lower resolution one, in order to rebuild protein coats for remodeled membranes of complex topology. The specific case of membrane remodeling by N-BAR domain containing proteins is considered here, although the overall method is general and thus applicable to other membrane remodeling phenomena. Our approach begins with a previously developed, discretized mesoscopic continuum membrane model (EM2) which has been shown to capture the reticulated membrane topologies often observed for N-BAR/liposome systems by electron microscopy (EM). The information in the EM2 model—directions of the local curvatures and a low resolution sample of the membrane surface—is then used to construct a coarse-grained (CG) system with one site per lipid and 26 sites per protein. We demonstrate the approach on pieces of EM2 structures with three different topologies that have been observed by EM: A tubule, a “Y” junction, and a torus. We show that the approach leads to structures that are stable under subsequent constant temperature CG simulation, and end by considering the future application of the methodology as a hybrid approach that combines experimental information with computer modeling.
Co-reporter:Shulu Feng and Gregory A. Voth
The Journal of Physical Chemistry B 2011 Volume 115(Issue 19) pp:5903-5912
Publication Date(Web):April 21, 2011
DOI:10.1021/jp2002194
Proton solvation properties and transport mechanisms have been studied in hydrated Nafion using the self-consistent multistate empirical valence bond (SCI-MS-EVB) method that includes the effects excess proton charge defect delocalization and Grotthuss proton hopping. It was found that sulfonate groups influence excess proton solvation, as well as the proton hydration structure, by stabilizing a more Zundel-like (H5O2+) structure in their first solvation shells. Hydrate proton-related hydrogen bond networks were observed to be more stable than networks with water alone. Diffusion rates, Arrhenius activation energies, and transport pathways were calculated and analyzed to characterize the nature of the proton transport. Diffusion rate analysis suggests that a proton-hopping mechanism dominates the proton transport for the studied water loading levels and that there is a clear degree of anticorrelation with the vehicular transport. The activation energy drops quickly with an increasing water content when the water loading level is smaller than ∼10 H2O/SO3–, which is consistent with experimental observations. The sulfonate groups were also found to affect the proton hopping directions. The temperature and water content effects on the proton transport pathways were also investigated.
Co-reporter:Ian F. Thorpe, David P. Goldenberg, and Gregory A. Voth
The Journal of Physical Chemistry B 2011 Volume 115(Issue 41) pp:11911-11926
Publication Date(Web):September 11, 2011
DOI:10.1021/jp204455g
Coarse-grained models can facilitate the efficient simulation of complex biological systems. In earlier studies the multiscale coarse-graining (MS-CG) method was employed to examine the folding landscape for two small peptides. In those studies, MS-CG force fields specific to each peptide were employed. We extend here the scope of that work with the goal of obtaining a transferable MS-CG force field which can be used to simulate the folded conformations of peptides with disparate structural motifs. Information obtained via MS-CG modeling was used to understand the characteristics of CG interactions which govern their capacity to be transferred between different peptide systems. We find that polar CG groups are least transferable in general, with interactions between CG sites representing the CO and NH groups on the peptide backbone being particularly resistant to facile transfer. Our results additionally suggest that, while there are limitations to the approach, the MS-CG method may provide a systematic path toward obtaining rigorously defined CG interactions with at least some degree of transferability. These studies also indicate that it may be possible to enhance the transferability of the MS-CG approach by identifying novel ways to combine information from different MS-CG force fields.
Co-reporter:Jianqing Xu, Yong Zhang, and Gregory A. Voth
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 2) pp:81-86
Publication Date(Web):December 23, 2010
DOI:10.1021/jz101536b
Reactive molecular dynamics simulations have been utilized to calculate the infrared (IR) spectra of acidic HCl solutions of varying concentration with the goal of achieving a better understanding of the spectral features of the hydrated excess protons in bulk water. To incorporate the essential physics of the hydrated proton, we carried out the simulations using the specialized self-consistent iterative multistate empirical valence bond (SCI-MS-EVB) method, which is a form of multiconfigurational (reactive) molecular dynamics. After the pure water absorption background was removed, the calculated difference spectra are in good agreement with prior experimental results. The continuous broad absorption band in the acidic IR spectrum is, for the first time, interpreted based on the concept of a dynamically distorted Eigen cation, H9O4+, which has been shown to provide the most accurate description for the charge defect character of the hydrated excess proton in liquid water.Keywords (keywords): Eigen; hydrated proton; infrared spectra; molecular dynamics; MS-EVB; proton solvation; Zundel;
Co-reporter:Zhen Cao ; Yuxing Peng ; Tianying Yan ; Shu Li ; Ailin Li
Journal of the American Chemical Society 2010 Volume 132(Issue 33) pp:11395-11397
Publication Date(Web):July 29, 2010
DOI:10.1021/ja1046704
A reactive molecular dynamics simulation employing the multistate empirical valence bond (MS-EVB) methodology is reported for the hydration structure of an excess proton in a (6,6) carbon nanotube as well as for the mechanism of proton transport (PT) within the nanoconfined environment. The proton is found to be hydrated in a distorted Zundel cation (H5O2+) form within the one-dimensional, confined water chain. Proton transfer events occur via a “Zundel−Zundel” mechanism through a transient H7O3+ intermediate that differs significantly from the “Eigen−Zundel−Eigen” mechanism found in bulk water.
Co-reporter:Kim F. Wong, Jason L. Sonnenberg, Francesco Paesani, Takeshi Yamamoto, Jiří Vaníček, Wei Zhang, H. Bernhard Schlegel, David A. Case, Thomas E. Cheatham III, William H. Miller, and Gregory A. Voth
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 9) pp:2566-2580
Publication Date(Web):August 5, 2010
DOI:10.1021/ct900579k
The rates of intramolecular proton transfer are calculated on a full-dimensional reactive electronic potential energy surface that incorporates high-level ab initio calculations along the reaction path and by using classical transition state theory, path-integral quantum transition state theory, and the quantum instanton approach. The specific example problem studied is malonaldehyde. Estimates of the kinetic isotope effect using the latter two methods are found to be in reasonable agreement with each other. Improvements and extensions of this practical, yet chemically accurate framework for the calculations of quantized, reactive dynamics are also discussed.
Co-reporter:Hanning Chen, Pu Liu, and Gregory A. Voth
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 10) pp:3039-3047
Publication Date(Web):August 31, 2010
DOI:10.1021/ct100318f
Nonbonded interactions between molecules usually include the van der Waals force and computationally expensive long-range electrostatic interactions. This article develops a more efficient approach: the effective-interaction multistate empirical-valence-bond (EI-MS-EVB) model. The EI-MS-EVB method relies on a mapping of all interactions onto a short-range and thus, computationally efficient effective potential. The effective potential is tabulated by matching its force to known trajectories obtained from the full-potential empirical multistate empirical-valence-bond (MS-EVB) model. The effective pairwise interaction depends on and is uniquely determined by the atomic configuration of the system, varying only with respect to the hydrogen-bonding topology. By comparing the EI-MS-EVB and full MS-EVB calculations of several equilibrium and dynamic properties important to hydrated excess proton solvation and transport, we show that the EI-MS-EVB model produces very accurate results for the specific system in which the tabulated potentials were generated. The EI-MS-EVB potential also transfers reasonably well to similar systems with different temperatures and box sizes. The EI-MS-EVB method also reduces the computational cost of the nonbonded interactions by about 1 order of magnitude in comparison with the full algorithm.
Co-reporter:Chris Knight, C. Mark Maupin, Sergei Izvekov, and Gregory A. Voth
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 10) pp:3223-3232
Publication Date(Web):September 23, 2010
DOI:10.1021/ct1004438
In this report, a general methodology is presented for the parametrization of a reactive force field using data from a condensed phase ab initio molecular dynamics (AIMD) simulation. This algorithm allows for the creation of an empirical reactive force field that accurately reproduces the underlying ab initio reactive surface while providing the ability to achieve long-time statistical sampling for large systems not possible with AIMD alone. In this work, a model for the hydrated excess proton is constructed where the hydronium cation and proton hopping portions of the model are statistically force-matched to the results of Car−Parrinello Molecular Dynamics (CPMD) simulations. The flexible nature of the algorithm also allows for the use of the more accurate classical simple point-charge flexible water (SPC/Fw) model to describe the water−water interactions while utilizing the ab initio data to create an overall multistate molecular dynamics (MS-MD) reactive model of the hydrated excess proton in water. The resulting empirical model for the system qualitatively reproduces thermodynamic and dynamic properties calculated from the ab initio simulation while being in good agreement with experimental results and previously developed multistate empirical valence bond (MS-EVB) models. The present methodology, therefore, bridges the AIMD technique with the MS-MD modeling of reactive events, while incorporating key strengths of both.
Co-reporter:Zhiyong Zhang and Gregory A. Voth
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 9) pp:2990-3002
Publication Date(Web):August 23, 2010
DOI:10.1021/ct100374a
High-resolution atomistic structures of many large biomolecular complexes have not yet been solved by experiments, such as X-ray crystallography or NMR. Often however low-resolution information is obtained by alternative techniques, such as cryo-electron microscopy or small-angle X-ray scattering. Coarse-grained (CG) models are an appropriate choice to computationally study these complexes given the limited resolution experimental data. One of the important questions therefore is how to define CG representations from these low-resolution density maps. This work provides a space-based essential dynamics coarse-graining (ED-CG) method to define a CG representation from a density map without detailed knowledge of its underlying atomistic structure and primary sequence information. This method is demonstrated on G-actin (both the atomic structure and its density map). It is then applied to the density maps of the Escherichia coli 70S ribosome and the microtubule. The results indicate that the method can define highly CG models that still preserve functionally important dynamics of large biomolecular complexes.
Co-reporter:Jim Pfaendtner;Enrique M. De La Cruz
PNAS 2010 Volume 107 (Issue 16 ) pp:7299-7304
Publication Date(Web):2010-04-20
DOI:10.1073/pnas.0911675107
We investigate, using molecular dynamics, how the severing protein, actin depolymerization factor (ADF)/cofilin, modulates
the structure, conformational dynamics, and mechanical properties of actin filaments. The actin and cofilactin filament bending
stiffness and corresponding persistence lengths obtained from all-atom simulations are comparable to values obtained from
analysis of thermal fluctuations in filament shape. Filament flexibility is strongly affected by the nucleotide-linked conformation
of the actin subdomain 2 DNase-I binding loop and the filament radial mass density distribution. ADF/cofilin binding between
subdomains 1 and 3 of a filament subunit triggers reorganization of subdomain 2 of the neighboring subunit such that the DNase-I
binding loop (DB-loop) moves radially away from the filament. Repositioning of the neighboring subunit DB-loop significantly
weakens subunit interactions along the long-pitch helix and lowers the filament bending rigidity. Lateral filament contacts
between the hydrophobic loop and neighboring short-pitch helix monomers in native filaments are also compromised with cofilin
binding. These works provide a molecular interpretation of biochemical solution studies documenting the disruption of filament
subunit interactions and also reveal the molecular basis of actin filament allostery and its linkage to ADF/cofilin binding.
Co-reporter:Sayan Bagchi, Dayton G. Thorpe, Ian F. Thorpe, Gregory A. Voth, and M. D. Fayer
The Journal of Physical Chemistry B 2010 Volume 114(Issue 51) pp:17187-17193
Publication Date(Web):December 3, 2010
DOI:10.1021/jp109203b
Myoglobin is an important protein for the study of structure and dynamics. Three conformational substates have been identified for the carbonmonoxy form of myoglobin (MbCO). These are manifested as distinct peaks in the IR absorption spectrum of the CO stretching mode. Ultrafast 2D IR vibrational echo chemical exchange experiments are used to observed switching between two of these substates, A1 and A3, on a time scale of <100 ps for two mutants of wild-type Mb. The two mutants are a single mutation of Mb, L29I, and a double mutation, T67R/S92D. Molecular dynamics (MD) simulations are used to model the structural differences between the substates of the two MbCO mutants. The MD simulations are also employed to examine the substate switching in the two mutants as a test of the ability of MD simulations to predict protein dynamics correctly for a system in which there is a well-defined transition over a significant potential barrier between two substates. For one mutant, L29I, the simulations show that translation of the His64 backbone may differentiate the two substates. The simulations accurately reproduce the experimentally observed interconversion time for the L29I mutant. However, MD simulations exploring the same His64 backbone coordinate fail to display substate interconversion for the other mutant, T67R/S92D, thus pointing to the likely complexity of the underlying protein interactions. We anticipate that understanding conformational dynamics in MbCO via ultrafast 2D IR vibrational echo chemical exchange experiments can help to elucidate fast conformational switching processes in other proteins.
Co-reporter:Yanting Wang and Gregory A. Voth
The Journal of Physical Chemistry B 2010 Volume 114(Issue 26) pp:8735-8743
Publication Date(Web):June 15, 2010
DOI:10.1021/jp1007768
The multiscale coarse-graining (MS-CG) method is used to construct solvent-free CG models for polyglutamine peptides having various repeat lengths. Because the resulting CG models have fewer degrees of freedom than a corresponding all-atom simulations, they make it possible to study the self-assembly of polyglutamines at high concentrations for the first time by allowing for better equilibration and statistical sampling that is well beyond the range achievable by all-atom models. Molecular dynamics (MD) simulations performed with these models show that polyglutamine monomers with repeat lengths ≤28 fluctuate between their folded and unfolded states. Monomers with 32 or more residues are stable and form α-helix solid structures. The degree of monomer compactness increases with chain length in both cases. CG MD simulations of equilibrium polyglutamine aggregates show that even at high concentrations, the system statistically fluctuates between heterogeneous and homogeneous configurations, rather than simply aggregates. The degree of aggregation and fluctuation increases with concentration and chain length. All of these phenomena are consistent with the experimental observations and may be explained by a mechanism that the collective nonbonded interactions between polyglutamine molecules in water solution are only weakly attractive. Finally, this work demonstrates that computer simulation of polypeptides self-assembly and aggregation, which is presently beyond the reach of all-atom MD simulations, is attainable using solvent-free MS-CG models.
Co-reporter:Aram Davtyan, Mijo Simunovic, Gregory A. Voth
Journal of Structural Biology (October 2016) Volume 196(Issue 1) pp:57-63
Publication Date(Web):1 October 2016
DOI:10.1016/j.jsb.2016.06.012
Protein-facilitated shape and topology changes of cell membranes are crucial for many biological processes, such as cell division, protein trafficking, and cell signaling. However, the inherently multiscale nature of membrane remodeling presents a considerable challenge for understanding the mechanisms and physics that drive this process. To address this problem, a multiscale approach that makes use of a diverse set of computational and experimental techniques is required. The atomistic simulations provide high-resolution information on protein-membrane interactions. Experimental techniques, like electron microscopy, on the other hand, resolve high-order organization of proteins on the membrane. Coarse-grained (CG) and mesoscale computational techniques provide the intermediate link between the two scales and can give new insights into the underlying mechanisms. In this Review, we present the recent advances in multiscale computational approaches established in our group. We discuss various CG and mesoscale approaches in studying the protein-mediated large-scale membrane remodeling.
Co-reporter:Yuxing Peng, Gregory A. Voth
Biochimica et Biophysica Acta (BBA) - Bioenergetics (April 2012) Volume 1817(Issue 4) pp:518-525
Publication Date(Web):April 2012
DOI:10.1016/j.bbabio.2011.11.017
Co-reporter:Chun-Liang Lai, Anand Srivastava, Carissa Pilling, Anna R. Chase, ... Gregory A. Voth
Journal of Molecular Biology (9 September 2013) Volume 425(Issue 17) pp:3073-3090
Publication Date(Web):9 September 2013
DOI:10.1016/j.jmb.2013.05.026
•Membrane docking reactions of the GRP1 PH domain are investigated.•Anionic phosphatidylserine lipids drive and accelerate the GRP1 PH domain membrane binding.•MD simulations quantitatively validate the EPR-determined membrane docking model.•MD shows the details at the binding interfaces and the stereospecific PIP3 binding.•Coarse-grained analysis reveals the dynamic character of the membrane-interacting loops.The pleckstrin homology (PH) domain of the general receptor of phosphoinositides 1 (GRP1) protein selectively binds to a rare signaling phospholipid, phosphatidylinositol (3,4,5)-trisphosphate (PIP3), in the membrane. The specific PIP3 lipid docking of GRP1 PH domain is essential to protein cellular function and is believed to occur in a stepwise process, electrostatic-driven membrane association followed by the specific PIP3 binding. By a combination of all-atom molecular dynamics (MD) simulations, coarse-grained analysis, electron paramagnetic resonance (EPR) membrane docking geometry, and fluorescence resonance energy transfer (FRET) kinetic studies, we have investigated the search and bind process in the GRP1 PH domain at the molecular scale. We simulated the two membrane binding states of the GRP1 PH domain in the PIP3 search process, before and after the GRP1 PH domain docks with the PIP3 lipid. Our results suggest that the background anionic phosphatidylserine lipids, which constitute around one-fifth of the membrane by composition, play a critical role in the initial stages of recruiting protein to the membrane surface through non-specific electrostatic interactions. Our data also reveal a previously unseen transient membrane association mechanism that is proposed to enable a two-dimensional “hopping” search of the membrane surface for the rare PIP3 target lipid. We further modeled the PIP3-bound membrane–protein system using the EPR membrane docking structure for the MD simulations, quantitatively validating the EPR membrane docking structure and augmenting our understanding of the binding interface with atomic-level detail. Several observations and hypotheses reached from our MD simulations are also supported by experimental kinetic studies.Download high-res image (416KB)Download full-size image
Co-reporter:Chun-Liang Lai, Christine C. Jao, Edward Lyman, Jennifer L. Gallop, ... Gregory A. Voth
Journal of Molecular Biology (9 November 2012) Volume 423(Issue 5) pp:800-817
Publication Date(Web):9 November 2012
DOI:10.1016/j.jmb.2012.08.010
Epsin possesses a conserved epsin N-terminal homology (ENTH) domain that acts as a phosphatidylinositol 4,5-bisphosphate‐lipid‐targeting and membrane‐curvature‐generating element. Upon binding phosphatidylinositol 4,5‐bisphosphate, the N-terminal helix (H0) of the ENTH domain becomes structured and aids in the aggregation of ENTH domains, which results in extensive membrane remodeling. In this article, atomistic and coarse-grained (CG) molecular dynamics (MD) simulations are used to investigate the structure and the stability of ENTH domain aggregates on lipid bilayers. EPR experiments are also reported for systems composed of different ENTH-bound membrane morphologies, including membrane vesicles as well as preformed membrane tubules. The EPR data are used to help develop a molecular model of ENTH domain aggregates on preformed lipid tubules that are then studied by CG MD simulation. The combined computational and experimental approach suggests that ENTH domains exist predominantly as monomers on vesiculated structures, while ENTH domains self-associate into dimeric structures and even higher‐order oligomers on the membrane tubes. The results emphasize that the arrangement of ENTH domain aggregates depends strongly on whether the local membrane curvature is isotropic or anisotropic. The molecular mechanism of ENTH‐domain-induced membrane vesiculation and tubulation and the implications of the epsin's role in clathrin-mediated endocytosis resulting from the interplay between ENTH domain membrane binding and ENTH domain self-association are also discussed.Download high-res image (272KB)Download full-size imageHighlights► ENTH domain plays an essential role in clathrin-mediated endocytosis. ► Membrane binding of ENTH domain is studied by a combined MD and EPR approach. ► A CG ENTH model for CG MD simulations is developed. ► ENTH-bound membrane tubules and vesicles are studied via CG MD simulations. ► The arrangement of ENTH domains highly correlates with local membrane curvature.
Co-reporter:Andrea Grafmüller, Gregory A. Voth
Structure (9 March 2011) Volume 19(Issue 3) pp:409-417
Publication Date(Web):9 March 2011
DOI:10.1016/j.str.2010.12.020
The complex polymerization dynamics of the microtubule (MT) plus end are closely linked to the hydrolysis of the GTP nucleotide bound to the β-tubulin. The destabilization is thought to be associated with the conformational change of the tubulin dimers from the straight conformation in the MT lattice to a curved conformation. It remains under debate whether this transformation is directly related to the nucleotide state, or a consequence of the longitudinal or lateral contacts in the MT lattice. Here, we present large-scale atomistic simulations of short tubulin protofilaments with both nucleotide states, starting from both extreme conformations. Our simulations indicate that both interdimer and intradimer contacts in both GDP and GTP-bound tubulin dimers and protofilaments in solution bend. There are no observable differences between the mesoscopic properties of the contacts in GTP and GDP-bound tubulin or the intradime and interdimer interfaces.Graphical AbstractDownload high-res image (341KB)Download full-size imageHighlights► GDP and GTP-bound tubulin protofilaments converge to a curved conformation ► No difference in mesoscopic properties of GDP and GTP filaments ► Rotation of the intermediate domain is not prevented by GTP ► Conformations resemble the 1SA2 structure closely with some additional contacts
Co-reporter:Hui Li, Hanning Chen, Christina Steinbronn, Binghua Wu, ... Gregory A. Voth
Journal of Molecular Biology (8 April 2011) Volume 407(Issue 4) pp:607-620
Publication Date(Web):8 April 2011
DOI:10.1016/j.jmb.2011.01.036
Prevention of cation permeation in wild-type aquaporin-1 (AQP1) is believed to be associated with the Asn-Pro-Ala (NPA) region and the aromatic/arginine selectivity filter (SF) domain. Previous work has suggested that the NPA region helps to impede proton permeation due to the protein backbone collective macrodipoles that create an environment favoring a directionally discontinuous channel hydrogen-bonded water chain and a large electrostatic barrier. The SF domain contributes to the proton permeation barrier by a spatial restriction mechanism and direct electrostatic interactions. To further explore these various effects, the free-energy barriers and the maximum cation conductance for the permeation of various cations through the AQP1-R195V and AQP1-R195S mutants are predicted computationally. The cations studied included the hydrated excess proton that utilizes the Grotthuss shuttling mechanism, a model “classical” charge localized hydronium cation that exhibits no Grotthuss shuttling, and a sodium cation. The hydrated excess proton was simulated using a specialized multi-state molecular dynamics method including a proper physical treatment of the proton shuttling and charge defect delocalization. Both AQP1 mutants exhibit a surprising cooperative effect leading to a reduction in the free-energy barrier for proton permeation around the NPA region due to altered water configurations in the SF region, with AQP1-R195S having a higher conductance than AQP1-R195V. The theoretical predictions are experimentally confirmed in wild-type AQP1 and the mutants expressed in Xenopus oocytes. The combined results suggest that the SF domain is a specialized structure that has evolved to impede proton permeation in aquaporins.Download high-res image (147KB)Download full-size imageResearch Highlights► Proton/cation permeation rates calculated in AQP1 mutants. ► Novel cooperative effect found in wild-type channel for blocking proton permeation. ► Computational predictions confirmed by electrophysiology experiments. ► Selectivity filter domain likely evolved to impede proton permeation in aquaporins.
Co-reporter:Mijo Simunovic, Carsten Mim, Thomas C. Marlovits, Guenter Resch, Vinzenz M. Unger, Gregory A. Voth
Biophysical Journal (6 August 2013) Volume 105(Issue 3) pp:
Publication Date(Web):6 August 2013
DOI:10.1016/j.bpj.2013.06.039
Key cellular processes are frequently accompanied by protein-facilitated shape changes in the plasma membrane. N-BAR-domain protein modules generate curvature by means of complex interactions with the membrane surface. The way they assemble and the mechanism by which they operate are largely dependent on their binding density. Although the mechanism at lower densities has recently begun to emerge, how membrane scaffolds form at high densities remains unclear. By combining electron microscopy and multiscale simulations, we show that N-BAR proteins at high densities can transform a lipid vesicle into a 3D tubular network. We show that this process is a consequence of excess adhesive energy combined with the local stiffening of the membrane, which occurs in a narrow range of mechanical properties of both the membrane and the protein. We show that lipid diffusion is significantly reduced by protein binding at this density regime and even more in areas of high Gaussian curvature, indicating a potential effect on molecular transport in cells. Finally, we reveal that the breaking of the bilayer topology is accompanied by the nematic arrangement of the protein on the surface, a structural motif that likely drives the formation of reticular structures in living cells.
Co-reporter:Gregory A. Voth
Biophysical Journal (5 February 2013) Volume 104(Issue 3) pp:
Publication Date(Web):5 February 2013
DOI:10.1016/j.bpj.2012.12.029
Co-reporter:Marissa G. Saunders, Gregory A. Voth
Structure (4 April 2012) Volume 20(Issue 4) pp:641-653
Publication Date(Web):4 April 2012
DOI:10.1016/j.str.2012.02.008
The interconversion of actin between monomeric and polymeric forms is a fundamental process in cell biology that is incompletely understood, in part because there is no high-resolution structure for filamentous actin. Several models have been proposed recently; identifying structural and dynamic differences between them is an essential step toward understanding actin dynamics. We compare three of these models, using coarse-grained analysis of molecular dynamics simulations to analyze the differences between them and evaluate their relative stability. Based on this analysis, we identify key motions that may be associated with polymerization, including a potential energetic barrier in the process. We also find that actin subunits are polymorphic; during simulations they assume a range of configurations remarkably similar to those seen in recent cryoEM images.Graphical AbstractDownload high-res image (258KB)Download full-size imageHighlights► Coarse-grained analysis of MD simulations is used to compare F-actin models ► After simulation, most subunits resemble the Oda model; strands are closer together ► Subunit flattening is coupled to a hinging motion of subdomain 2 ► After equilibration, F-actin models differ mainly in the position of subdomain 2
Co-reporter:Gary S. Ayton, Gregory A. Voth
Biophysical Journal (3 November 2010) Volume 99(Issue 9) pp:
Publication Date(Web):3 November 2010
DOI:10.1016/j.bpj.2010.08.018
Multiscale computer simulations, employing a combination of experimental data and coarse-graining methods, are used to explore the structure of the immature HIV-1 virion. A coarse-grained (CG) representation is developed for the virion membrane shell and Gag polypeptides using molecular level information. Building on the results from electron cryotomography experiments, the simulations under certain conditions reveal the existence of an incomplete p6 hexameric lattice formed from hexameric bundles of the Gag CA domains. In particular, the formation and stability of the immature Gag lattice at the CG level requires enhanced interfacial interactions of the CA protein C-terminal domains (CTDs). An exact mapping of the CG representation back to the molecular level then allows for detailed atomistic molecular dynamics studies to confirm the existence of these enhanced CACTD interactions and to probe their possible origin. The multiscale simulations further provide insight into potential CACTD mutations that may disrupt or modify the Gag immature lattice assembly process in the immature HIV-1 virion.
Co-reporter:Andrea Grafmüller, Eva G. Noya, Gregory A. Voth
Journal of Molecular Biology (26 June 2013) Volume 425(Issue 12) pp:2232-2246
Publication Date(Web):26 June 2013
DOI:10.1016/j.jmb.2013.03.029
► Nucleotide (NT)-dependent conformations are observed for several loops in β-tubulin in dimers. ► Similar NT-dependent changes are found in a piece of tubulin lattice. ► The lattice contacts stabilize NT-dependent loop conformations also in α-tubulin. ► Coarse-grained analysis predicts stronger interactions in guanosine-triphosphate-bound tubulin.Microtubule (MT) stability is related to the hydrolysis of the guanosine triphosphate nucleotide (NT) bound to β-tubulin. However, the molecular mechanism by which the NT state influences the stability of the contacts in the MT lattice remains elusive. Here, we present large-scale atomistic simulations of different tubulin aggregates, including individual dimers, short protofilaments, a small lattice patch, and a piece of the MT lattice with two infinite protofilaments in both NT states. Together with a coarse-grained (CG) analysis of the fluctuations, these simulations highlight several regions of the protein where local changes are induced by the NT state or by the lateral and longitudinal contacts in the aggregates. Additionally, the CG analysis provides an indication of how the structural changes affect the bonds between the proteins. The results suggest a consistent picture of a possible molecular mechanism by which the NT state induces changes in the H1–S2 loop and more stable longitudinal bonds, both of which locate the H1–S2 and M-loop in more favorable positions to form lateral contacts.Download high-res image (167KB)Download full-size image
Co-reporter:Andrea Grafmüller, Eva G. Noya, Gregory A. Voth
Journal of Molecular Biology (26 June 2013) Volume 425(Issue 12) pp:2232-2246
Publication Date(Web):26 June 2013
DOI:10.1016/j.jmb.2013.03.029
► Nucleotide (NT)-dependent conformations are observed for several loops in β-tubulin in dimers. ► Similar NT-dependent changes are found in a piece of tubulin lattice. ► The lattice contacts stabilize NT-dependent loop conformations also in α-tubulin. ► Coarse-grained analysis predicts stronger interactions in guanosine-triphosphate-bound tubulin.Microtubule (MT) stability is related to the hydrolysis of the guanosine triphosphate nucleotide (NT) bound to β-tubulin. However, the molecular mechanism by which the NT state influences the stability of the contacts in the MT lattice remains elusive. Here, we present large-scale atomistic simulations of different tubulin aggregates, including individual dimers, short protofilaments, a small lattice patch, and a piece of the MT lattice with two infinite protofilaments in both NT states. Together with a coarse-grained (CG) analysis of the fluctuations, these simulations highlight several regions of the protein where local changes are induced by the NT state or by the lateral and longitudinal contacts in the aggregates. Additionally, the CG analysis provides an indication of how the structural changes affect the bonds between the proteins. The results suggest a consistent picture of a possible molecular mechanism by which the NT state induces changes in the H1–S2 loop and more stable longitudinal bonds, both of which locate the H1–S2 and M-loop in more favorable positions to form lateral contacts.Download high-res image (167KB)Download full-size image
Co-reporter:Edward Lyman, Haosheng Cui, Gregory A. Voth
Biophysical Journal (22 September 2010) Volume 99(Issue 6) pp:
Publication Date(Web):22 September 2010
DOI:10.1016/j.bpj.2010.06.074
Many cellular processes require the generation of highly curved regions of cell membranes by interfacial membrane proteins. A number of such proteins are now known, and several mechanisms of curvature generation have been suggested, but so far a quantitative understanding of the importance of the various potential mechanisms remains elusive. Following previous theoretical work, we consider the electrostatic attraction that underlies the scaffold mechanism of membrane bending in the context of the N-BAR domain of amphiphysin. Analysis of atomistic molecular dynamics simulations reveals considerable water between the membrane and the positively charged concave face of the BAR, even when it is tightly bound to highly curved membranes. This results in significant screening of electrostatic interactions, suggesting that electrostatic attraction is not the main driving force behind curvature sensing, supporting recent experimental work. These results also emphasize the need for care when building coarse-grained models of protein-membrane interactions. These results are emphasized by simulations of oligomerized amphiphysin N-BARs at the atomistic and coarse-grained level. In the coarse-grained simulations, we find a strong dependence of the induced curvature on the dielectric screening.
Co-reporter:Haosheng Cui, Carsten Mim, Francisco X. Vázquez, Edward Lyman, Vinzenz M. Unger, Gregory A. Voth
Biophysical Journal (22 January 2013) Volume 104(Issue 2) pp:
Publication Date(Web):22 January 2013
DOI:10.1016/j.bpj.2012.12.006
Endophilin N-BAR (N-terminal helix and Bin/amphiphysin/Rvs) domain tubulates and vesiculates lipid membranes in vitro via its crescent-shaped dimer and four amphipathic helices that penetrate into membranes as wedges. Like F-BAR domains, endophilin N-BAR also forms a scaffold on membrane tubes. Unlike F-BARs, endophilin N-BARs have N-terminal H0 amphipathic helices that are proposed to interact with other N-BARs in oligomer lattices. Recent cryo-electron microscopy reconstructions shed light on the organization of the N-BAR lattice coats on a nanometer scale. However, because of the resolution of the reconstructions, the precise positioning of the amphipathic helices is still ambiguous. In this work, we applied a coarse-grained model to study various membrane remodeling scenarios induced by endophilin N-BARs. We found that H0 helices of N-BARs prefer to align in an antiparallel manner at two ends of the protein to form a stable lattice. The deletion of H0 helices causes disruption of the lattice. In addition, we analyzed the persistence lengths of the protein-coated tubes and found that the stiffness of endophilin N-BAR-coated tubules qualitatively agrees with previous experimental work studying N-BAR-coated tubules. Large-scale simulations on membrane liposomes revealed a systematic relation between H0 helix density and local membrane curvature fluctuations. The data also suggest that the H0 helix is required for BARs to form organized structures on the liposome, further illustrating its important function.
Co-reporter:Francisco X. Vázquez, Vinzenz M. Unger, Gregory A. Voth
Biophysical Journal (22 January 2013) Volume 104(Issue 2) pp:
Publication Date(Web):22 January 2013
DOI:10.1016/j.bpj.2012.12.009
Endophilin is a key protein involved in clathrin-mediated endocytosis. Previous computational and experimental work suggested that the N-terminal helix is embedded into the membrane to induce curvature; however, the role of the SH3 domain remains controversial. To address this issue, we performed computer simulations of the endophilin dimer in solution to understand the interaction between the N-BAR and SH3 domains and its effect on biological function. We predict that the helix binds to the SH3 domain through hydrophobic and salt-bridge interactions. This protects the hydrophobic residues on both domains and keeps the SH3 domain near the end of the N-BAR domain, in agreement with previous experimental results. The complex has a binding strength similar to a few hydrogen bonds (13.0 ± 0.6 kcal/mol), and the SH3 domain stabilizes the structure of the N-terminal helix in solution. Electrostatic calculations show a large region of strongly positive electrostatic potential near the N-terminal that can orient the helix toward the membrane and likely embed the helix into the membrane surface. This predicted mechanism suggests that endophilin can select for both curvature and electrostatic potential when interacting with membranes, highlighting the importance of the SH3 domain in regulating the function of endophilin.
Co-reporter:Haosheng Cui, Edward Lyman, Gregory A. Voth
Biophysical Journal (2 March 2011) Volume 100(Issue 5) pp:
Publication Date(Web):2 March 2011
DOI:10.1016/j.bpj.2011.01.036
There are several examples of membrane-associated protein domains that target curved membranes. This behavior is believed to have functional significance in a number of essential pathways, such as clathrin-mediated endocytosis, which involve dramatic membrane remodeling and require the recruitment of various cofactors at different stages of the process. This work is motivated in part by recent experiments that demonstrated that the amphipathic N-terminal helix of endophilin (H0) targets curved membranes by binding to hydrophobic lipid bilayer packing defects which increase in number with increasing membrane curvature. Here we use state-of-the-art atomistic simulation to explore the packing defect structure of curved membranes, and the effect of this structure on the folding of H0. We find that not only are packing defects increased in number with increasing membrane curvature, but also that their size distribution depends nontrivially on the curvature, falling off exponentially with a decay constant that depends on the curvature, and crucially that even on highly curved membranes defects large enough to accommodate the hydrophobic face of H0 are never observed. We furthermore find that a percolation model for the defects explains the defect size distribution, which implies that larger defects are formed by coalescence of noninteracting smaller defects. We also use the recently developed metadynamics algorithm to study in detail the effect of such defects on H0 folding. It is found that the comparatively larger defects found on a convex membrane promote H0 folding by several kcal/mol, while the smaller defects found on flat and concave membrane surfaces inhibit folding by kinetically trapping the peptide. Together, these observations suggest H0 folding is a cooperative process in which the folding peptide changes the defect structure relative to an unperturbed membrane.
Co-reporter:Mijo Simunovic, Gregory A. Voth
Biophysical Journal (18 July 2012) Volume 103(Issue 2) pp:
Publication Date(Web):18 July 2012
DOI:10.1016/j.bpj.2012.06.018
Hsp90, the most abundant cellular protein, has been implicated in numerous physiological and pathological processes. It controls protein folding and prevents aggregation, but it also plays a role in cancer and neurological disorders, making it an attractive drug target. Experimental efforts have demonstrated its remarkable structural flexibility and conformational complexity, which enable it to accommodate a variety of clients, but have not been able to provide a detailed molecular description of the conformational transitions. In our molecular dynamics simulations, Hsp90 underwent dramatic structural rearrangements into energetically favorable stretched and compact states. The transitions were guided by key electrostatic interactions between specific residues of opposite subunits. Nucleotide-bound structures showed the same conformational flexibility, although ADP and ATP seemed to potentiate these interactions by stabilizing two different closed conformations. Our observations may explain the difference in dynamic behavior observed among Hsp90 homologs, and the atomic resolution of the conformational transitions helps elucidate the complex chaperone machinery.
Co-reporter:Chun-Liang Lai, Kyle E. Landgraf, Gregory A. Voth, Joseph J. Falke
Journal of Molecular Biology (17 September 2010) Volume 402(Issue 2) pp:301-310
Publication Date(Web):17 September 2010
DOI:10.1016/j.jmb.2010.07.037
Protein kinase Cα (PKCα) possesses a conserved C2 domain (PKCα C2 domain) that acts as a Ca2+-regulated membrane targeting element. Upon activation by Ca2+, the PKCα C2 domain directs the kinase protein to the plasma membrane, thereby stimulating an array of cellular pathways. At sufficiently high Ca2+ concentrations, binding of the C2 domain to the target lipid phosphatidylserine (PS) is sufficient to drive membrane association; however, at typical physiological Ca2+ concentrations, binding to both PS and phosphoinositidyl-4,5-bisphosphate (PIP2) is required for specific plasma membrane targeting. Recent EPR studies have revealed the membrane docking geometries of the PKCα C2 domain docked to (i) PS alone and (ii) both PS and PIP2 simultaneously. These two EPR docking geometries exhibit significantly different tilt angles relative to the plane of the membrane, presumably induced by the large size of the PIP2 headgroup. The present study utilizes the two EPR docking geometries as starting points for molecular dynamics simulations that investigate atomic features of the protein-membrane interaction. The simulations yield approximately the same PIP2-triggered change in tilt angle observed by EPR. Moreover, the simulations predict a PIP2:C2 stoichiometry approaching 2:1 at a high PIP2 mole density. Direct binding measurements titrating the C2 domain with PIP2 in lipid bilayers yield a 1:1 stoichiometry at moderate mole densities and a saturating 2:1 stoichiometry at high PIP2 mole densities. Thus, the experiment confirms the target lipid stoichiometry predicted by EPR-guided molecular dynamics simulations. Potential biological implications of the observed docking geometries and PIP2 stoichiometries are discussed.
Co-reporter:John M.A. Grime, Gregory A. Voth
Biophysical Journal (17 October 2012) Volume 103(Issue 8) pp:
Publication Date(Web):17 October 2012
DOI:10.1016/j.bpj.2012.09.007
The early stages in the formation of the HIV-1 capsid (CA) protein lattice are investigated. The underlying coarse-grained (CG) model is parameterized directly from experimental data and examined under various native contact interaction strengths, CA dimer interfacial configurations, and local surface curvatures. The mechanism of early contiguous mature-style CA p6 lattice formation is explored, and a trimer-of-dimers structure is found to be crucial for CA lattice production. Quasi-equivalent generation of both the pentamer and hexamer components of the HIV-1 viral CA is also demonstrated, and the formation of pentamers is shown to be highly sensitive to local curvature, supporting the view that such inclusions in high-curvature regions allow closure of the viral CA surface. The complicated behavior of CA lattice self-assembly is shown to be reducible to a relatively simple function of the trimer-of-dimers behavior.
Co-reporter:Yong Zhang, Gregory A. Voth
Biophysical Journal (16 November 2011) Volume 101(Issue 10) pp:
Publication Date(Web):16 November 2011
DOI:10.1016/j.bpj.2011.10.021
Using a reactive molecular dynamics simulation methodology, the free energy barrier for water-mediated proton transport between the two proton gating residues Glu203 and Glu148 in the ClC-ec1 antiporter, including the Grotthuss mechanism of proton hopping, was calculated. Three different chloride-binding states, with 1), both the central and internal Cl−, 2), the central Cl− only, and 3), the internal Cl− only, were considered and the coupling to the H+ transport studied. The results show that both the central and internal Cl− are essential for the proton transport from Glu203 to Glu148 to have a favorite free energy driving force. The rotation of the Glu148 side chain was also found to be independent of the internal chloride binding state. These results emphasize the importance of the 2:1 stoichiometry of this well-studied Cl−/H+ antiporter.
Co-reporter:Marissa G. Saunders, Gregory A. Voth
Journal of Molecular Biology (14 October 2011) Volume 413(Issue 1) pp:279-291
Publication Date(Web):14 October 2011
DOI:10.1016/j.jmb.2011.07.068
In the monomeric actin crystal structure, the positions of a highly organized network of waters are clearly visible within the active site. However, the recently proposed models of filamentous actin (F-actin) did not extend to including these waters. Since the water network is important for ATP hydrolysis, information about water position is critical to understanding the increased rate of catalysis upon filament formation. Here, we show that waters in the active site are essential for intersubdomain rotational flexibility and that they organize the active-site structure. Including the crystal structure waters during simulation setup allows us to observe distinct changes in the active-site structure upon the flattening of the actin subunit, as proposed in the Oda model for F-actin. We identify changes in both protein position and water position relative to the phosphate tail that suggest a mechanism for accelerating the rate of nucleotide hydrolysis in F-actin by stabilizing charge on the β-phosphate and by facilitating deprotonation of catalytic water.Download high-res image (252KB)Download full-size imageResearch Highlights► Upon filament formation, ATP hydrolysis by actin is accelerated by a factor of 104. ► Molecular dynamics simulations of F-actin filaments suggest a mechanism for this increase. ► Waters in the active site stabilize the filament and organize the active site. ► Subunit flattening rearranges the active-site water network around the γ-phosphate. ► These waters help stabilize the β-phosphate and deprotonate the catalytic water.
Co-reporter:Jun Fan, Marissa G. Saunders, Esmael J. Haddadian, Karl F. Freed, ... Gregory A. Voth
Journal of Molecular Biology (12 April 2013) Volume 425(Issue 7) pp:1225-1240
Publication Date(Web):12 April 2013
DOI:10.1016/j.jmb.2013.01.020
The actin regulatory protein cofilin plays a central role in actin assembly dynamics by severing filaments and increasing the concentration of ends from which subunits add and dissociate. Cofilin binding modifies the average structure and mechanical properties of actin filaments, thereby promoting fragmentation of partially decorated filaments at boundaries of bare and cofilin-decorated segments. Despite extensive evidence for cofilin-dependent changes in filament structure and mechanics, it is unclear how the two processes are linked at the molecular level. Here, we use molecular dynamics simulations and coarse-grained analyses to evaluate the molecular origins of the changes in filament compliance due to cofilin binding. Filament subunits with bound cofilin are less flat and maintain a significantly more open nucleotide cleft than bare filament subunits. Decorated filament segments are less twisted, thinner (considering only actin), and less connected than their bare counterparts, which lowers the filament bending persistence length and torsional stiffness. Using coarse-graining as an analysis method reveals that cofilin binding increases the average distance between the adjacent long-axis filament subunit, thereby weakening their interaction. In contrast, a fraction of lateral filament subunit contacts are closer and presumably stronger with cofilin binding. A cofilactin interface contact identified by cryo-electron microscopy is unstable during simulations carried out at 310 K, suggesting that this particular interaction may be short lived at ambient temperatures. These results reveal the molecular origins of cofilin-dependent changes in actin filament mechanics that may promote filament severing.Download high-res image (150KB)Download full-size imageHighlights► Cofilin binding increases actin filament flexibility. ► Cofilin binding opens the nucleotide-binding cleft of actin subunits. ► Cofilin weakens longitudinal subunit contacts and strengthens lateral contacts. ► Cofilactin intersubunit contacts have reduced density and cross-sectional area. ► Reorganization of longitudinal contacts appears to regulate filament flexibility.
Co-reporter:Jim Pfaendtner, Niels Volkmann, Dorit Hanein, Paul Dalhaimer, ... Gregory A. Voth
Journal of Molecular Biology (10 February 2012) Volume 416(Issue 1) pp:148-161
Publication Date(Web):10 February 2012
DOI:10.1016/j.jmb.2011.12.025
We investigated the structure, properties and dynamics of the actin filament branch junction formed by actin-related protein (Arp) 2/3 complex using all-atom molecular dynamics (MD) simulations based on a model fit to a reconstruction from electron tomograms. Simulations of the entire structure consisting of 31 protein subunits together with solvent molecules containing ∼ 3 million atoms were performed for an aggregate time of 175 ns. One 75-ns simulation of the original reconstruction was compared to two 50-ns simulations of alternate structures, showing that the hypothesized branch junction structure is very stable. Our simulations revealed that the interface between Arp2/3 complex and the mother actin filament features a large number of salt bridges and hydrophobic contacts, many of which are dynamic and formed/broken on the timescale of the simulation. The simulations suggest that the DNase binding loops in Arp3, and possibly Arp2, form stabilizing contacts with the mother filament. Unbiased comparison of models sampled from the MD simulation trajectory with the primary experimental electron tomography data identified regions were snapshots from the simulation provide atomic details of the model structures and also pinpoints regions where the initial modeling based on the electron tomogram reconstruction may be suboptimal.Download high-res image (98KB)Download full-size imageHighlights► We use MD simulation to study the structure and dynamics of the Arp2/3 branch junction. ► We compare two different filament models and study two different models for the active Arp2/3 complex. ► We study the atomic scale features of the actin–Arp2/3 binding interface. ► We performed unbiased comparison of models sampled from the MD simulation trajectory with the primary experimental electron tomography data to pinpoint regions where the initial modeling based on the electron tomogram reconstruction may be suboptimal.
Co-reporter:Joseph L. Baker, Gregory A. Voth
Biophysical Journal (1 October 2013) Volume 105(Issue 7) pp:
Publication Date(Web):1 October 2013
DOI:10.1016/j.bpj.2013.08.023
Actin and myosin interact with one another to perform a variety of cellular functions. Central to understanding the processive motion of myosin on actin is the characterization of the individual states along the mechanochemical cycle. We present an all-atom molecular dynamics simulation of the myosin II S1 domain in the rigor state interacting with an actin filament. We also study actin-free myosin in both rigor and post-rigor conformations. Using all-atom level and coarse-grained analysis methods, we investigate the effects of myosin binding on actin, and of actin binding on myosin. In particular, we determine the domains of actin and myosin that interact strongly with one another at the actomyosin interface using a highly coarse-grained level of resolution, and we identify a number of salt bridges and hydrogen bonds at the interface of myosin and actin. Applying coarse-grained analysis, we identify differences in myosin states dependent on actin-binding, or ATP binding. Our simulations also indicate that the actin propeller twist-angle and nucleotide cleft-angles are influenced by myosin at the actomyosin interface. The torsional rigidity of the myosin-bound filament is also calculated, and is found to be increased compared to previous simulations of the free filament.
Co-reporter:Takefumi Yamashita
Journal of the American Chemical Society () pp:
Publication Date(Web):December 19, 2011
DOI:10.1021/ja209176e
Cytochrome c oxidase (CcO), known as complex IV of the electron transport chain, plays several important roles in aerobic cellular respiration. Electrons transferred from cytochrome c to CcO’s catalytic site reduce molecular oxygen and produce a water molecule. These electron transfers also drive active proton pumping from the matrix (N-side) to intermembrane region (P-side) in mitochondria; the resultant proton gradient activates ATP synthase to produce ATP from ADP. Although the existence of the coupling between the electron transfer and the proton transport (PT) is established experimentally, its mechanism is not yet fully understood at the molecular level. In this work, it is shown why the reduction of heme a is essential for proton pumping. This is demonstrated via novel reactive molecular dynamics (MD) simulations that can describe the Grotthuss shuttling associated with the PT as well as the dynamic delocalization of the excess proton electronic charge defect. Moreover, the “valve” role of the Glu242 residue (bovine CcO notation) and the gate role of d-propionate of heme a3 (PRDa3) in the explicit PT are explicitly demonstrated for the first time. These results provide conclusive evidence for the CcO proton transporting mechanism inferred from experiments, while deepening the molecular level understanding of the CcO proton switch.
Co-reporter:Edward Lyman, Haosheng Cui and Gregory A. Voth
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 22) pp:NaN10436-10436
Publication Date(Web):2011/04/18
DOI:10.1039/C0CP02978E
We present a method for “inverse coarse graining,” rebuilding a higher resolution model from a lower resolution one, in order to rebuild protein coats for remodeled membranes of complex topology. The specific case of membrane remodeling by N-BAR domain containing proteins is considered here, although the overall method is general and thus applicable to other membrane remodeling phenomena. Our approach begins with a previously developed, discretized mesoscopic continuum membrane model (EM2) which has been shown to capture the reticulated membrane topologies often observed for N-BAR/liposome systems by electron microscopy (EM). The information in the EM2 model—directions of the local curvatures and a low resolution sample of the membrane surface—is then used to construct a coarse-grained (CG) system with one site per lipid and 26 sites per protein. We demonstrate the approach on pieces of EM2 structures with three different topologies that have been observed by EM: A tubule, a “Y” junction, and a torus. We show that the approach leads to structures that are stable under subsequent constant temperature CG simulation, and end by considering the future application of the methodology as a hybrid approach that combines experimental information with computer modeling.

![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6.png)
![3,5,8-Trioxa-4-phosphahexacos-17-en-1-aminium,4-hydroxy-N,N,N-trimethyl-9-oxo-7-[[(1-oxohexadecyl)oxy]methyl]-, inner salt,4-oxide, (7R,17Z)-](http://img.cochemist.com/ccimg/26900/26853-31-6_b.png)
![3,5,9-Trioxa-4-phosphatricosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxotetradecyl)oxy]-, inner salt, 4-oxide](http://img.cochemist.com/ccimg/18700/18656-38-7.png)
![3,5,9-Trioxa-4-phosphatricosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxotetradecyl)oxy]-, inner salt, 4-oxide](http://img.cochemist.com/ccimg/18700/18656-38-7_b.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)



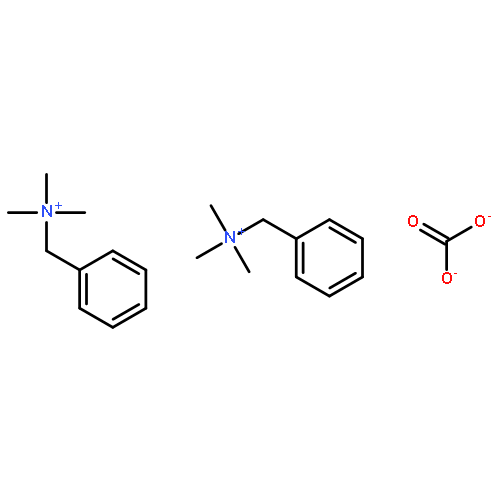


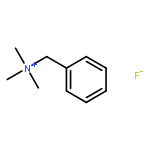




![(6R,9AR)-OCTAHYDRO-2H-PYRIDO[1,2-A]PYRAZIN-6-YLMETHANOL](http://img.cochemist.com/ccimg/13700/13699-48-4.png)
![(6R,9AR)-OCTAHYDRO-2H-PYRIDO[1,2-A]PYRAZIN-6-YLMETHANOL](http://img.cochemist.com/ccimg/13700/13699-48-4_b.png)




![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1.png)
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1_b.png)




![3,5,9-Trioxa-4-phosphaheneicosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxododecyl)oxy]-, inner salt, 4-oxide,(7R)-](http://img.cochemist.com/ccimg/18200/18194-25-7.png)
![3,5,9-Trioxa-4-phosphaheneicosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxododecyl)oxy]-, inner salt, 4-oxide,(7R)-](http://img.cochemist.com/ccimg/18200/18194-25-7_b.png)


![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4_b.png)





