Co-reporter:Qiao Wang, Xiaochuan Ma, Junli Jia and Hao Fei
Journal of Materials Chemistry A 2016 vol. 4(Issue 8) pp:1535-1543
Publication Date(Web):26 Jan 2016
DOI:10.1039/C5TB02783G
Liposomes are extensively used as drug carriers because of their biocompatibility, low toxicity, and controlled release properties, however challenges exist in the control of their particle size, surface properties and targeting functionality. In this work, we report a peptide–lipid nanoparticle platform that can achieve nanoparticle formation, surface functionalization and hydrophobic drug loading in an integrated assembly process. A designer peptide that harbors bivalent amphipathic α-helices linked by a central loop (ALA peptide) was used to encapsulate lipid nanoparticles (LNPs). The bivalency design affords higher peptide helicity and lipid-packaging efficiency, and allows encapsulated hydrophobic molecules for more stability under long-term storage. The central loop structure displays sufficient surface exposure as demonstrated by the interaction between penta-histidine installed LNPs and Ni-NTA agarose. RGD-inserted and cytotoxic iridium complex-encapsulated LNPs showed preferential entry and selective cytotoxicity to integrin high expression cancer cells, while showing reduced toxicity to non-cancer cells. Further study indicates that a constrained cyclic conformation of RGD is required to fully exert targeting capability, suggesting an intact structural exposure on the LNP surface. In summary, we demonstrate a simple yet effective method of peptide-based LNP surface modification with potential for various targeted deliveries of hydrophobic drugs.
Co-reporter:Xiaoli Liu; Rui Cao; Sha Wang; Junli Jia
Journal of Medicinal Chemistry 2016 Volume 59(Issue 11) pp:5238-5247
Publication Date(Web):May 19, 2016
DOI:10.1021/acs.jmedchem.5b02016
Cationic amphipathic peptides (CAPs) are known to be able to cause membrane destabilization and induce cell death, yet how the hydrophobicity, amphipathicity, and lysine (K)/arginine (R) composition synergistically affect the peptide activity remains incompletely understood. Here, we designed a panel of peptides based on the well-known anticancer peptide KLA. Increasing hydrophobicity enhanced the cytotoxicities of both the K- and R-rich peptides. Peptides with an intact amphipathic helical interface can cause instant cell death through a membrane lysis mechanism. Interestingly, rearranging the residue positions to minimize amphipathicity caused a great decrease of cytotoxicity to the K-rich peptides but not to the R-rich peptides. The amphipathicity-minimized R-rich peptide 6 (RL2) (RLLRLLRLRRLLRL-NH2) penetrated the cell membrane and induced caspase-3-dependent apoptotic cell death. We found that the modulation of hydrophobicity, amphipathicity, and K/R residues leads to distinct mechanisms of action of cationic hydrophobic peptides. Amphipathicity-reduced, arginine-rich cationic hydrophobic peptides (CHPs) may represent a new class of peptide therapeutics.
Co-reporter:Rui Cao ; Junli Jia ; Xiaochuan Ma ; Ming Zhou
Journal of Medicinal Chemistry 2013 Volume 56(Issue 9) pp:3636-3644
Publication Date(Web):April 17, 2013
DOI:10.1021/jm4001665
The cellular behavior and toxicity effect of organometallic complexes depend largely on their peripheral ligands. In this study, we have synthesized a series of novel luminescent cationic iridium(III) complexes by tuning the ancillary N∧N ligand based on a structure [Ir(ppy)2(N∧N)]+ (ppy = 1-phenyl-pyridine; N∧N = 2,2′-bipyridine (bpy, 1) or phenanthroline (phen, 2) or 4,7-diphenyl-1,10- phenanthroline (DIP, 3)). As the size of coordinated N∧N ligand increases, absorbance/emission efficiency, quantum yields, lipophilicity, and cell uptake rates of the complexes also increase, in a general order: 3 > 2 > 1. All three complexes display anticancer activity, with 3 exhibiting the highest cellular uptake efficiency and the greatest cytotoxic activities in several cancer cell lines with IC50s lower than that of cisplatin. Because of its strong hydrophobic nature, the death inducer 3 was found to accumulate favorably to endoplasmic reticulum (ER) and to cause ER stress in cells. The fast cytosolic release of calcium from stressed ER disturbed the morphology and function of mitochondria, initiating an intrinsic apoptotic pathway. Understanding of the cell death mechanism would help further structure–activity optimization on these novel Ir(III) complexes as emerging cancer therapeutics.
Co-reporter:Xiaochuan Ma;Xiaobo Wang;Ming Zhou
Advanced Healthcare Materials 2013 Volume 2( Issue 12) pp:1638-1643
Publication Date(Web):
DOI:10.1002/adhm.201300037
Abstract
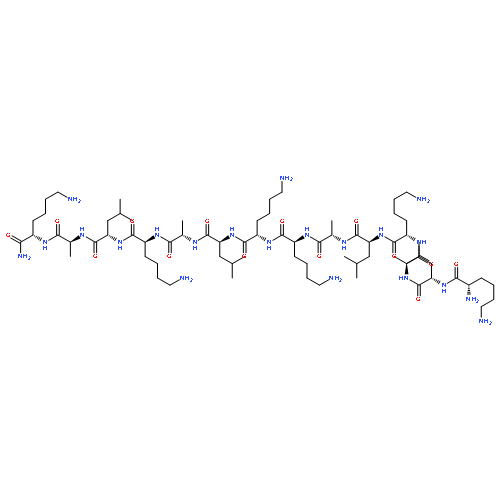
Design and construction of multifunctional nanoparticles for effective delivery and therapeutic application remains a challenging task. It is desirable that nanoparticles can overcome multiple biological barriers and reach specific cellular locations to achieve maximum therapeutic effects. This aim often requires the fine tuning of nanoparticles' chemical and physical properties, as well as better understanding of their interaction with live cells. A peptide-modified gold–nanoparticle platform is designed, which consists of a 20-nm gold core stabilized with a layer of biotinylated CALNN-based peptides and a further layer of tetrameric streptavidins for functionalization with biotinylated molecules. The nanoassembly undergoes an efficient dynamin-dependent and caveolae-mediated endocytosis pathway, and displays highly specific localization to mitochondria, organelles of great therapeutic importance. When functionalized with a cytotoxic peptide (KLA: (KLAKLAK)2), the KLA-anchored nanoassembly exhibits dramatically enhanced anticancer activity, thousands of times stronger than that of the free KLA peptide, likely because of its improved cell entry efficiency, mitochondria-specific delivery, and the polyvalent effect of the nanoassembly. The study opens up the possibility of developing mitochondria-targeted nanomedicines.
Co-reporter:Xiaobo Wang;Junli Jia;Zezhu Huang;Dr. Ming Zhou;Dr. Hao Fei
Chemistry - A European Journal 2011 Volume 17( Issue 29) pp:8028-8032
Publication Date(Web):
DOI:10.1002/chem.201100568
Co-reporter:Xiaochuan Ma ; Junli Jia ; Rui Cao ; Xiaobo Wang
Journal of the American Chemical Society () pp:
Publication Date(Web):December 8, 2014
DOI:10.1021/ja511656q
In the field of peptide drug discovery, structural constraining and fluorescent labeling are two sought-after techniques important for both basic research and pharmaceutical development. In this work, we describe an easy-to-use approach for simultaneous peptide cyclization and luminescent labeling based on iridium(III)–histidine coordination (Ir–HH cyclization). Using a series of model peptides with histidine flanking each terminus, the binding activity and reaction kinetics of Ir–HH cyclization of different ring sizes were characterized. In the series, Ir–HAnH (n = 2, 3) with moderate ring sizes provides appropriate flexibility and proper distance between histidines for cyclic formation, which leads to the best binding affinity and structural stability in physiological conditions, as compared to other Ir–HH-cyclized peptides with smaller (n = 0, 1) or larger (n = 4, 5) ring sizes. Ir–HRGDH, an Ir–HH-cyclized peptide containing integrin targeting motif Arg-Gly-Asp (RGD), showed better targeting affinity than its linear form and enhanced membrane permeability in comparison with fluorescein-labeled cyclic RGDyK peptide. Cell death inducing peptide KLA-linked Ir–HRGDH (Ir–HRGDH–KLA) showed dramatically enhanced cytotoxicity and high selectivity for cancer cells versus noncancer cells. These data demonstrate that the method conveniently combines structural constraining of peptides with luminescent imaging capabilities, which facilitates functional and intracellular characterization of potential peptide-based drug leads, thus introducing a new tool to meet emerging needs in medicinal research.
Co-reporter:Qiao Wang, Xiaochuan Ma, Junli Jia and Hao Fei
Journal of Materials Chemistry A 2016 - vol. 4(Issue 8) pp:NaN1543-1543
Publication Date(Web):2016/01/26
DOI:10.1039/C5TB02783G
Liposomes are extensively used as drug carriers because of their biocompatibility, low toxicity, and controlled release properties, however challenges exist in the control of their particle size, surface properties and targeting functionality. In this work, we report a peptide–lipid nanoparticle platform that can achieve nanoparticle formation, surface functionalization and hydrophobic drug loading in an integrated assembly process. A designer peptide that harbors bivalent amphipathic α-helices linked by a central loop (ALA peptide) was used to encapsulate lipid nanoparticles (LNPs). The bivalency design affords higher peptide helicity and lipid-packaging efficiency, and allows encapsulated hydrophobic molecules for more stability under long-term storage. The central loop structure displays sufficient surface exposure as demonstrated by the interaction between penta-histidine installed LNPs and Ni-NTA agarose. RGD-inserted and cytotoxic iridium complex-encapsulated LNPs showed preferential entry and selective cytotoxicity to integrin high expression cancer cells, while showing reduced toxicity to non-cancer cells. Further study indicates that a constrained cyclic conformation of RGD is required to fully exert targeting capability, suggesting an intact structural exposure on the LNP surface. In summary, we demonstrate a simple yet effective method of peptide-based LNP surface modification with potential for various targeted deliveries of hydrophobic drugs.

![Iridium(1 ), diaquabis[2-(2-pyridinyl-κN)phenyl-κC]-, (OC-6-33)-, 1,1,1-trifluoromethanesulfonate (1:1)](/data/chemimg/107100/153297-46-2.png)
![Iridium(1 ), diaquabis[2-(2-pyridinyl-κN)phenyl-κC]-, (OC-6-33)-, 1,1,1-trifluoromethanesulfonate (1:1)](/data/chemimg/107100/153297-46-2_b.png)


![4-(4'-Methyl-[2,2'-bipyridin]-4-yl)butanoic acid](http://img.cochemist.com/ccimg/114600/114527-28-5.png)
![4-(4'-Methyl-[2,2'-bipyridin]-4-yl)butanoic acid](http://img.cochemist.com/ccimg/114600/114527-28-5_b.png)