Co-reporter:Narendran G-DayanandanEric W. Scocchera, Santosh Keshipeddy, Heather F. Jones, Amy C. Anderson, Dennis L. Wright
Organic Letters 2017 Volume 19(Issue 1) pp:142-145
Publication Date(Web):December 13, 2016
DOI:10.1021/acs.orglett.6b03438
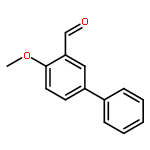
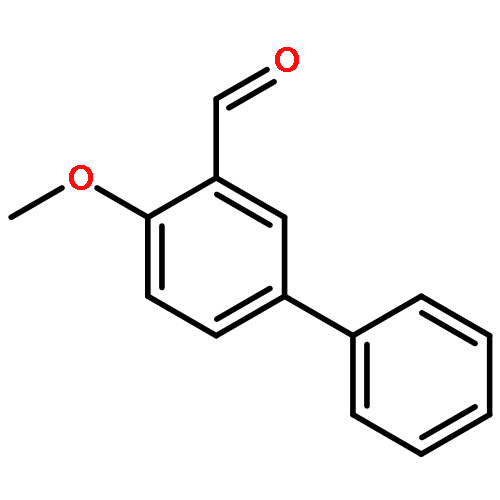
To develop next generation antifolates for the treatment of trimethoprim-resistant bacteria, synthetic methods were needed to prepare a diverse array of 3-aryl-propynes with various substitutions at the propargyl position. A direct route was sought whereby nucleophilic addition of acetylene to aryl carboxaldehydes would be followed by reduction or substitution of the resulting propargyl alcohol. The direct reduction, methylation, and dimethylation of these readily available alcohols provide efficient access to this uncommon functional array. In addition, an unusual silane exchange reaction was observed in the reduction of the propargylic alcohols.
Co-reporter:Stephanie M. Reeve; Eric Scocchera; Jacob J. Ferreira; Narendran G-Dayanandan; Santosh Keshipeddy; Dennis L. Wright;Amy C. Anderson
Journal of Medicinal Chemistry 2016 Volume 59(Issue 13) pp:6493-6500
Publication Date(Web):June 16, 2016
DOI:10.1021/acs.jmedchem.6b00688
Drug-resistant enzymes must balance catalytic function with inhibitor destabilization to provide a fitness advantage. This sensitive balance, often involving very subtle structural changes, must be achieved through a selection process involving a minimal number of eligible point mutations. As part of a program to design propargyl-linked antifolates (PLAs) against trimethoprim-resistant dihydrofolate reductase (DHFR) from Staphylococcus aureus, we have conducted a thorough study of several clinically observed chromosomal mutations in the enzyme at the cellular, biochemical, and structural levels. Through this work, we have identified a promising lead series that displays significantly greater activity against these mutant enzymes and strains than TMP. The best inhibitors have enzyme inhibition and MIC values near or below that of trimethoprim against wild-type S. aureus. Moreover, these studies employ a series of crystal structures of several mutant enzymes bound to the same inhibitor; analysis of the structures reveals a more detailed molecular understanding of drug resistance in this important enzyme.
Co-reporter:Eric Scocchera, Stephanie M. Reeve, Santosh Keshipeddy, Michael N. Lombardo, Behnoush Hajian, Adrienne E. Sochia, Jeremy B. Alverson, Nigel D. Priestley, Amy C. Anderson, and Dennis L. Wright
ACS Medicinal Chemistry Letters 2016 Volume 7(Issue 7) pp:692
Publication Date(Web):May 5, 2016
DOI:10.1021/acsmedchemlett.6b00120
Although classical, negatively charged antifolates such as methotrexate possess high affinity for the dihydrofolate reductase (DHFR) enzyme, they are unable to penetrate the bacterial cell wall, rendering them poor antibacterial agents. Herein, we report a new class of charged propargyl-linked antifolates that capture some of the key contacts common to the classical antifolates while maintaining the ability to passively diffuse across the bacterial cell wall. Eight synthesized compounds exhibit extraordinary potency against Gram-positive S. aureus with limited toxicity against mammalian cells and good metabolic profile. High resolution crystal structures of two of the compounds reveal extensive interactions between the carboxylate and active site residues through a highly organized water network.Keywords: antifolate; Dihydrofolate reductase; Escherichia coli; methotrexate; MRSA; Staphylococcus aureus; trimethoprim
Co-reporter:Santosh Keshipeddy; Stephanie M. Reeve; Amy C. Anderson
Journal of the American Chemical Society 2015 Volume 137(Issue 28) pp:8983-8990
Publication Date(Web):June 22, 2015
DOI:10.1021/jacs.5b01442
While antifolates such as Bactrim (trimethoprim-sulfamethoxazole; TMP-SMX) continue to play an important role in treating community-acquired methicillin-resistant Staphylococcus aureus (CA-MRSA), resistance-conferring mutations, specifically F98Y of dihydrofolate reductase (DHFR), have arisen and compromise continued use. In an attempt to extend the lifetime of this important class, we have developed a class of propargyl-linked antifolates (PLAs) that exhibit potent inhibition of the enzyme and bacterial strains. Probing the role of the configuration at the single propargylic stereocenter in these inhibitors required us to develop a new approach to nonracemic 3-aryl-1-butyne building blocks by the pairwise use of asymmetric conjugate addition and aldehyde dehydration protocols. Using this new route, a series of nonracemic PLA inhibitors was prepared and shown to possess potent enzyme inhibition (IC50 values <50 nM), antibacterial effects (several with MIC values <1 μg/mL) and to form stable ternary complexes with both wild-type and resistant mutants. Unexpectedly, crystal structures of a pair of individual enantiomers in the wild-type DHFR revealed that the single change in configuration of the stereocenter drove the selection of an alternative NADPH cofactor, with the minor α-anomer appearing with R-27. Remarkably, this cofactor switching becomes much more prevalent when the F98Y mutation is present. The observation of cofactor site plasticity leads to a postulate for the structural basis of TMP resistance in DHFR and also suggests design strategies that can be used to target these resistant enzymes.
Co-reporter:Michael D. VanHeyst
European Journal of Organic Chemistry 2015 Volume 2015( Issue 7) pp:1387-1401
Publication Date(Web):
DOI:10.1002/ejoc.201403116
Abstract
Isolated from the marine sponge Dysidea frondosa, the frondosin family of meroterpenoid natural products were shown to be antagonists of the binding of the cytokine interleukin-8 to its receptor. With implications of interleukin-8 management being associated with a wide range of acute and chronic inflammatory disorders, as well as tumor progression and/or metathesis, there have been a multitude of syntheses of these natural products in the hopes of developinging new pharmacological agents. However, assignment of the absolute configurations of these natural products became unclear, because the first two syntheses of frondosin B, by Danishefsky and by Trauner, produced conflicting assignments for the single stereogenic center at C8. Here we describe the initial stereochemical mystery surrounding the Danishefsky and Trauner syntheses and provide evidence through ensuing syntheses of frondosins B and A that resolve this issue. These subsequent asymmetric syntheses have established that (1) the natural product occurs as the R enantiomer, and (2) a late-stage stereochemical inversion occurred during the Trauner and Wright syntheses of frondosin B.
Co-reporter:E.Zachary Oblak ; Michael D. VanHeyst ; Jin Li ; Andrew J. Wiemer
Journal of the American Chemical Society 2014 Volume 136(Issue 11) pp:4309-4315
Publication Date(Web):February 27, 2014
DOI:10.1021/ja413106t
The asymmetric total syntheses of the natural products (+)- and (−)-frondosin B and (+)-frondosin A are reported based on a diastereoselective cycloaddition between tetrabromocyclopropene and an annulated furan to provide a highly functionalized common building block. The bridged bicyclic intermediate could be stereo- and chemoselectively manipulated to produce the two structurally distinct members of the frondosins. Both syntheses feature regioselective palladium-coupling reactions and an unprecedented phosphine-mediated ether bridge cleavage. Surprisingly, the planned enantioselective synthesis of frondosin B led to the opposite epimer of the natural product, suggesting an unusual late stage stereoinversion at C8. Frondosin A, but not frondosin B, was shown to have selective antiproliferative activity against several B-cell lines.
Co-reporter:Narendran G-Dayanandan ; Janet L. Paulsen ; Kishore Viswanathan ; Santosh Keshipeddy ; Michael N. Lombardo ; Wangda Zhou ; Kristen M. Lamb ; Adrienne E. Sochia ; Jeremy B. Alverson ; Nigel D. Priestley ; Dennis L. Wright ;Amy C. Anderson
Journal of Medicinal Chemistry 2014 Volume 57(Issue 6) pp:2643-2656
Publication Date(Web):February 25, 2014
DOI:10.1021/jm401916j
Species of Candida, primarily C. albicans and with increasing prevalence, C. glabrata, are responsible for the majority of fungal bloodstream infections that cause morbidity, especially among immune compromised patients. While the development of new antifungal agents that target the essential enzyme, dihydrofolate reductase (DHFR), in both Candida species would be ideal, previous attempts have resulted in antifolates that exhibit inconsistencies between enzyme inhibition and antifungal properties. In this article, we describe the evaluation of pairs of propargyl-linked antifolates that possess similar physicochemical properties but different shapes. All of these compounds are effective at inhibiting the fungal enzymes and the growth of C. glabrata; however, the inhibition of the growth of C. albicans is shape-dependent with extended para-linked compounds proving more effective than compact, meta-linked compounds. Using crystal structures of DHFR from C. albicans and C. glabrata bound to lead compounds, 13 new para-linked compounds designed to inhibit both species were synthesized. Eight of these compounds potently inhibit the growth of both fungal species with three compounds displaying dual MIC values less than 1 μg/mL. Analysis of the active compounds shows that shape and distribution of polar functionality is critical in achieving dual antifungal activity.
Co-reporter:Sophia N. Ononye, Michael D. VanHeyst, E. Zachary Oblak, Wangda Zhou, Mohamed Ammar, Amy C. Anderson, and Dennis L. Wright
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 8) pp:757-761
Publication Date(Web):June 10, 2013
DOI:10.1021/ml400158k
Natural products have long been recognized as a rich source of potent therapeutics but further development is often limited by high structural complexity and high molecular weight. In contrast, at the core of the thujaplicins is a lead-like tropolone scaffold characterized by relatively low molecular weight, ample sites for diversification, and metal-binding functionality poised for targeting a range of metalloenzyme drug targets. Here, we describe the development of this underutilized scaffold for the discovery of tropolone derivatives that function as isozyme-selective inhibitors of the validated anticancer drug target, histone deacetylase (HDAC). Several monosubstituted tropolones display remarkable levels of selectivity for HDAC2 and potently inhibit the growth of T-cell lymphocyte cell lines. The tropolones represent a new chemotype of isozyme-selective HDAC inhibitors.Keywords: HDAC; isozyme-selectivity; metalloenzyme; T-lymphocyte cancer cell lines; thujaplicin; Tropolone;
Co-reporter:Wangda Zhou, Eric W. Scocchera, Dennis L. Wright and Amy C. Anderson
MedChemComm 2013 vol. 4(Issue 6) pp:908-915
Publication Date(Web):25 Apr 2013
DOI:10.1039/C3MD00104K
Over the past six decades, the folate biosynthetic pathway has provided a rich source of drug targets for the treatment of proliferative diseases. Drugs targeting dihydrofolate reductase have been especially successful as anticancer (methotrexate), antibacterial (trimethoprim, TMP) and antiprotozoal (cycloguanil, pyrimethamine) therapeutics. While trimethoprim remains a clinically important antimicrobial DHFR inhibitor, resistance by point mutations in otherwise sensitive strains as well as the natural insensitivity of several species limits its use. In this review, an historical overview of the attempts to develop drugs that target the folate pathway is presented along with a discussion of the basis of insensitivity to trimethoprim. From this vantage, we have developed the propargyl-linked antifolates as potent inhibitors of TMP-insensitive enzymes and strains. The structural basis of the increased affinity is detailed to promote the development of further generations of antifolates.
Co-reporter:Janet L. Paulsen, Kishore Viswanathan, Dennis L. Wright, Amy C. Anderson
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 5) pp:1279-1284
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmcl.2013.01.008
A novel strategy for targeting the pathogenic organisms Candida albicans and Candida glabrata focuses on the development of potent and selective antifolates effective against dihydrofolate reductase. Crystal structure analysis suggested that an essential loop at the active site (Thr 58-Phe 66) differs from the analogous residues in the human enzyme, potentially providing a mechanism for achieving selectivity. In order to probe the role of this loop, we employed chemical synthesis, crystal structure determination and molecular dynamics simulations. The results of these analyses show that the loop residues undergo ligand-induced conformational changes that are similar among the fungal and human species.
Co-reporter:Michael D. VanHeyst, E. Zachary Oblak, and Dennis L. Wright
The Journal of Organic Chemistry 2013 Volume 78(Issue 20) pp:10555-10559
Publication Date(Web):September 18, 2013
DOI:10.1021/jo4017502
An improved methodology for the preparation of enantiopure oxabicyclo[3.2.1]octadienes via a stereodivergent resolution is reported. High catalyst control proximal to the oxabridged stereocenter produces readily separable diastereomers in high yield (>92%) and with excellent optical purity (>95% ee). This resolution strategy is amenable to large-scale preparations, and the utility of the resolution was further demonstrated in the asymmetric preparation of a key intermediate used in the synthesis of the antibiotic (−)-platensimycin.
Co-reporter:E. Zachary Oblak, Erin S. D. Bolstad, Sophia N. Ononye, Nigel D. Priestley, M. Kyle Hadden and Dennis L. Wright
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 43) pp:8597-8604
Publication Date(Web):24 Sep 2012
DOI:10.1039/C2OB26553B
A direct route to analogs of the naturally occurring tropolone β-thujaplicin has been developed in just four steps from furan. Using this method, a series of derivatives were synthesized and evaluated. Several of these compounds demonstrated very high levels of potency against bacterial and fungal pathogens with good selectivity over mammalian cells.
Co-reporter:Kishore Viswanathan, Dennis J. Hoover, Jeannie Hwang, Meagan L. Wisniewski, Uzoma S. Ikonne, Ben A. Bahr, and Dennis L. Wright
ACS Medicinal Chemistry Letters 2012 Volume 3(Issue 11) pp:920
Publication Date(Web):September 9, 2012
DOI:10.1021/ml300197h
Lysosomes are involved in protein turnover and removing misfolded species, and their enzymes have the potential to offset the defect in proteolytic clearance that contributes to the age-related dementia Alzheimer's disease (AD). The weak cathepsin B and L inhibitor Z-Phe-Ala-diazomethylketone (PADK) enhances lysosomal cathepsin levels at low concentrations, thereby eliciting protective clearance of PHF-τ and Aβ42 in the hippocampus and other brain regions. Here, a class of positive modulators is established with compounds decoupled from the cathepsin inhibitory properties. We utilized PADK as a departure point to develop nonpeptidic structures with the hydroxyethyl isostere. The first-in-class modulators SD1002 and SD1003 exhibit improved levels of cathepsin up-regulation but almost complete removal of cathepsin inhibitory properties as compared to PADK. Isomers of the lead compound SD1002 were synthesized, and the modulatory activity was determined to be stereoselective. In addition, the lead compound was tested in transgenic mice with results indicating protection against AD-type protein accumulation pathology.Keywords: Alzheimer's disease; cathepsin B; lysosomal modulator; nonpeptidic inhibitors; PADK
Co-reporter:Kishore Viswanathan, Sophia N. Ononye, Harold D. Cooper, M. Kyle Hadden, Amy C. Anderson, Dennis L. Wright
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 22) pp:6919-6922
Publication Date(Web):15 November 2012
DOI:10.1016/j.bmcl.2012.09.015
Naturally occurring furanosteroids such as viridin and wortmannin have long been known as potent inhibitors of the lipid kinase PI-3K. We have been interested in directly accessing analogs of these complex natural products from abundant steroid feedstock materials. In this communication, we describe the synthesis of viridin/wortmannin hybrid molecules from readily available building blocks that function as PI-3K inhibitors and maintain their electrophilic properties. The compounds also show anti-proliferative effects against a breast cancer line.
Co-reporter:E. Zachary Oblak and Dennis L. Wright
Organic Letters 2011 Volume 13(Issue 9) pp:2263-2265
Publication Date(Web):March 31, 2011
DOI:10.1021/ol2005775
A stereocontrolled approach to a key platensimycin intermediate was achieved from a commercially available furylcarboxylate. Key to our approach is the highly efficient formal [4 + 3] cyclocondensation of a substituted furan with tetrabromocyclopropene along with an intramolecular γ-alkylation to construct the final ring of the caged intermediate.
Co-reporter:Yanzhong Zhang, E. Zachary Oblak, Erin S.D. Bolstad, Amy C. Anderson, Jerry P. Jasinski, Ray J. Butcher, Dennis L. Wright
Tetrahedron Letters 2010 Volume 51(Issue 47) pp:6120-6122
Publication Date(Web):24 November 2010
DOI:10.1016/j.tetlet.2010.09.058
The natural product liphagal has been shown to function as a reasonably potent and selective inhibitor of the key signaling enzyme PI-3Kα. We have been interested in developing an analog class of PI-3K inhibitors based upon this unusual terpenoid natural product. Toward that end, we have evaluated the binding of the natural product to its target protein computationally and formulated a class of simplified analogs based on the structural analysis. Utilizing the cycloadduct derived from tetrabromocyclopropene and furan, we were able to generate a key, versatile scaffold upon which to pursue this analog design.
Co-reporter:Jieying Liu;David B. Bolstad;Adrienne E. Smith;Nigel D. Priestley;Amy C. Anderson
Chemical Biology & Drug Design 2009 Volume 73( Issue 1) pp:62-74
Publication Date(Web):
DOI:10.1111/j.1747-0285.2008.00745.x
Candida glabrata, a fungal strain resistant to many commonly administered antifungal agents, has become an emerging threat to human health. In previous work, we validated that the essential enzyme, dihydrofolate reductase, is a drug target in C. glabrata. Using a crystal structure of dihydrofolate reductase from C. glabrata bound to an initial lead compound, we designed a class of biphenyl antifolates that potently and selectively inhibit both the enzyme and the growth of the fungal culture. In this work, we explore the structure–activity relationships of this class of antifolates with four new high resolution crystal structures of enzyme:inhibitor complexes and the synthesis of four new inhibitors. The designed inhibitors are intended to probe key hydrophobic pockets visible in the crystal structure. The crystal structures and an evaluation of the new compounds reveal that methyl groups at the meta and para positions of the distal phenyl ring achieve the greatest number of interactions with the pathogenic enzyme and the greatest degree of selectivity over the human enzyme. Additionally, antifungal activity can be tuned with substitution patterns at the propargyl and para-phenyl positions.
Co-reporter:Jieying Liu, David B. Bolstad, Adrienne E. Smith, Nigel D. Priestley, Dennis L. Wright, Amy C. Anderson
Chemistry & Biology 2008 Volume 15(Issue 9) pp:990-996
Publication Date(Web):22 September 2008
DOI:10.1016/j.chembiol.2008.07.013
Candida glabrata is a lethal fungal pathogen resistant to many antifungal agents and has emerged as a critical target for drug discovery. Over the past several years, we have been developing a class of propargyl-linked antifolates as antimicrobials and hypothesized that these compounds could be effective inhibitors of dihydrofolate reductase (DHFR) from C. glabrata. We initially screened a small collection of these inhibitors and found modest levels of potency. Subsequently, we determined the crystal structure of C. glabrata DHFR bound to a representative inhibitor with data to 1.6 Å resolution. Using this structure, we designed and synthesized second-generation inhibitors. These inhibitors bind the C. glabrata DHFR enzyme with subnanomolar potency, display greater than 2000-fold levels of selectivity over the human enzyme, and inhibit the growth of C. glabrata at levels observed with clinically employed therapeutics.
Co-reporter:David B. Bolstad ; Erin S. D. Bolstad ; Kathleen M. Frey ; Dennis L. Wright ;Amy C. Anderson
Journal of Medicinal Chemistry 2008 Volume 51(Issue 21) pp:6839-6852
Publication Date(Web):October 4, 2008
DOI:10.1021/jm8009124
Cryptosporidiosis is an emerging infectious disease that can be life-threatening in an immune-compromised individual and causes gastrointestinal distress lasting up to 2 weeks in an immune-competent individual. There are few therapeutics available for effectively treating this disease. We have been exploring dihydrofolate reductase (DHFR) as a potential target in Cryptosporidium. On the basis of the structure of the DHFR enzyme from C. hominis, we have developed a novel scaffold that led to the discovery of potent (38 nM) and efficient inhibitors of this enzyme. Recently, we have advanced these inhibitors to the next stage of development. Using the structures of both the protozoal and human enzymes, we have developed inhibitors with nanomolar potency (1.1 nM) against the pathogenic enzyme and high levels (1273-fold) of selectivity over the human enzyme.
Co-reporter:Jennifer M. Beierlein ; Kathleen M. Frey ; David B. Bolstad ; Phillip M. Pelphrey ; Tammy M. Joska ; Adrienne E. Smith ; Nigel D. Priestley ; Dennis L. Wright ;Amy C. Anderson
Journal of Medicinal Chemistry 2008 Volume 51(Issue 23) pp:7532-7540
Publication Date(Web):November 13, 2008
DOI:10.1021/jm800776a
Bacillus anthracis, the causative agent of anthrax, poses a significant biodefense danger. Serious limitations in approved therapeutics and the generation of resistance have produced a compelling need for new therapeutic agents against this organism. Bacillus anthracis is known to be insensitive to the clinically used antifolate, trimethoprim, because of a lack of potency against the dihydrofolate reductase enzyme. Herein, we describe a novel lead series of B. anthracis dihydrofolate reductase inhibitors characterized by an extended trimethoprim-like scaffold. The best lead compound adds only 22 Da to the molecular weight and is 82-fold more potent than trimethoprim. An X-ray crystal structure of this lead compound bound to B. anthracis dihydrofolate reductase in the presence of NADPH was determined to 2.25 Å resolution. The structure reveals several features that can be exploited for further development of this lead series.
Co-reporter:E. Zachary Oblak, Erin S. D. Bolstad, Sophia N. Ononye, Nigel D. Priestley, M. Kyle Hadden and Dennis L. Wright
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 43) pp:NaN8604-8604
Publication Date(Web):2012/09/24
DOI:10.1039/C2OB26553B
A direct route to analogs of the naturally occurring tropolone β-thujaplicin has been developed in just four steps from furan. Using this method, a series of derivatives were synthesized and evaluated. Several of these compounds demonstrated very high levels of potency against bacterial and fungal pathogens with good selectivity over mammalian cells.

![L-Alaninamide,N-[(phenylmethoxy)carbonyl]-L-valyl-N-[(1S)-1-(carboxymethyl)-3-fluoro-2-oxopropyl]-](http://img.cochemist.com/ccimg/161500/161401-82-7.png)
![L-Alaninamide,N-[(phenylmethoxy)carbonyl]-L-valyl-N-[(1S)-1-(carboxymethyl)-3-fluoro-2-oxopropyl]-](http://img.cochemist.com/ccimg/161500/161401-82-7_b.png)


















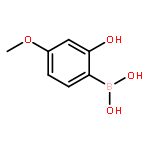
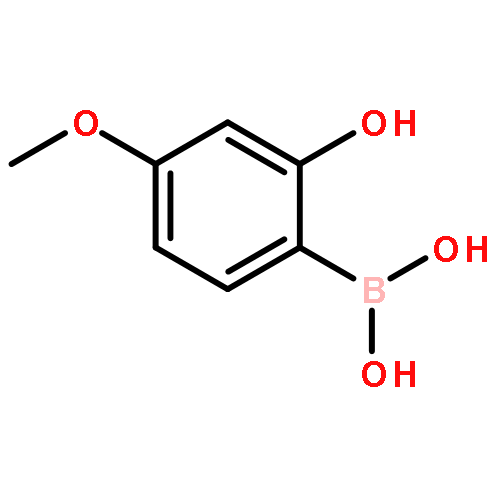
![1H-Benzo[b]benzo[6,7]cyclohepta[1,2-d]furan-9-carboxaldehyde, 2,3,4,4a,5,6,7,12c-octahydro-10,11-dihydroxy-4,4,7,12c-tetramethyl-, (4aS,7R,12cS)-](/data/chemimg/109600/874292-32-7.png)
![1H-Benzo[b]benzo[6,7]cyclohepta[1,2-d]furan-9-carboxaldehyde, 2,3,4,4a,5,6,7,12c-octahydro-10,11-dihydroxy-4,4,7,12c-tetramethyl-, (4aS,7R,12cS)-](/data/chemimg/109600/874292-32-7_b.png)


![8-Oxabicyclo[3.2.1]octa-3,6-dien-2-one, 3,4-dibromo-, (1R,5S)-](http://img.cochemist.com/ccimg/777900/777864-91-2.png)
![8-Oxabicyclo[3.2.1]octa-3,6-dien-2-one, 3,4-dibromo-, (1R,5S)-](http://img.cochemist.com/ccimg/777900/777864-91-2_b.png)
![8-OXABICYCLO[3.2.1]OCTA-3,6-DIEN-2-ONE, 3,4-DIBROMO-, (1S,5R)-](http://img.cochemist.com/ccimg/777900/777864-90-1.png)
![8-OXABICYCLO[3.2.1]OCTA-3,6-DIEN-2-ONE, 3,4-DIBROMO-, (1S,5R)-](http://img.cochemist.com/ccimg/777900/777864-90-1_b.png)


![8-Oxabicyclo[3.2.1]octa-3,6-dien-2-ol, 3,4-dibromo-, (1R,2S,5S)-rel-](http://img.cochemist.com/ccimg/657400/657389-18-9.png)
![8-Oxabicyclo[3.2.1]octa-3,6-dien-2-ol, 3,4-dibromo-, (1R,2S,5S)-rel-](http://img.cochemist.com/ccimg/657400/657389-18-9_b.png)


![Ethanone, 1-(4-methoxy[1,1'-biphenyl]-3-yl)-](http://img.cochemist.com/ccimg/412100/412027-31-7.png)
![Ethanone, 1-(4-methoxy[1,1'-biphenyl]-3-yl)-](http://img.cochemist.com/ccimg/412100/412027-31-7_b.png)




![3-Buten-2-ol, 1-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, (2R)-](http://img.cochemist.com/ccimg/191900/191800-37-0.png)
![3-Buten-2-ol, 1-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, (2R)-](http://img.cochemist.com/ccimg/191900/191800-37-0_b.png)




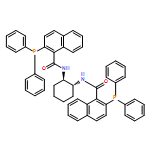
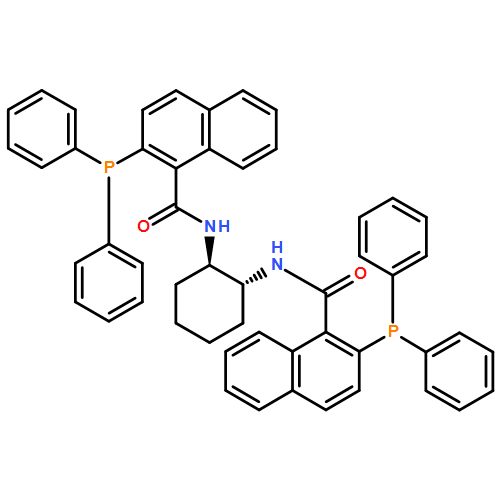
![Ferrocene,1-[(1S)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2S)-](http://img.cochemist.com/ccimg/162300/162291-02-3.png)
![Ferrocene,1-[(1S)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2S)-](http://img.cochemist.com/ccimg/162300/162291-02-3_b.png)
![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2R)-](http://img.cochemist.com/ccimg/155900/155806-35-2.png)
![Ferrocene,1-[(1R)-1-(dicyclohexylphosphino)ethyl]-2-(diphenylphosphino)-, (2R)-](http://img.cochemist.com/ccimg/155900/155806-35-2_b.png)
![L-Proline,N-[[(2S,3S)-3-[(propylamino)carbonyl]-2-oxiranyl]carbonyl]-L-isoleucyl-, methylester](http://img.cochemist.com/ccimg/147900/147859-80-1.png)
![L-Proline,N-[[(2S,3S)-3-[(propylamino)carbonyl]-2-oxiranyl]carbonyl]-L-isoleucyl-, methylester](http://img.cochemist.com/ccimg/147900/147859-80-1_b.png)


![Dimethyl(2-methyl-2-propanyl)[(tributylstannyl)methoxy]silane](http://img.cochemist.com/ccimg/123100/123061-64-3.png)
![Dimethyl(2-methyl-2-propanyl)[(tributylstannyl)methoxy]silane](http://img.cochemist.com/ccimg/123100/123061-64-3_b.png)
![3-Buten-2-ol, 1-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, (2S)-](http://img.cochemist.com/ccimg/91400/91376-49-7.png)
![3-Buten-2-ol, 1-[[(1,1-dimethylethyl)diphenylsilyl]oxy]-, (2S)-](http://img.cochemist.com/ccimg/91400/91376-49-7_b.png)
![bicyclo[2.2.1]hepta-2,5-diene,4-diphenylphosphanylbutyl(diphenyl)phosphane,rhodium,tetrafluoroborate](http://img.cochemist.com/ccimg/82500/82499-43-2.png)
![bicyclo[2.2.1]hepta-2,5-diene,4-diphenylphosphanylbutyl(diphenyl)phosphane,rhodium,tetrafluoroborate](http://img.cochemist.com/ccimg/82500/82499-43-2_b.png)
![Carbamic acid,N-[(1S)-2-[[(1S)-3-diazo-1-methyl-2-oxopropyl]amino]-2-oxo-1-(phenylmethyl)ethyl]-,phenylmethyl ester](http://img.cochemist.com/ccimg/71800/71732-53-1.png)
![Carbamic acid,N-[(1S)-2-[[(1S)-3-diazo-1-methyl-2-oxopropyl]amino]-2-oxo-1-(phenylmethyl)ethyl]-,phenylmethyl ester](http://img.cochemist.com/ccimg/71800/71732-53-1_b.png)
![Carbamic acid,N-[(1S)-2-[[(1S)-3-diazo-2-oxo-1-(phenylmethyl)propyl]amino]-2-oxo-1-(phenylmethyl)ethyl]-,phenylmethyl ester](http://img.cochemist.com/ccimg/65200/65178-14-5.png)
![Carbamic acid,N-[(1S)-2-[[(1S)-3-diazo-2-oxo-1-(phenylmethyl)propyl]amino]-2-oxo-1-(phenylmethyl)ethyl]-,phenylmethyl ester](http://img.cochemist.com/ccimg/65200/65178-14-5_b.png)


![Ethanone, 1-(3-methoxy[1,1'-biphenyl]-4-yl)-](http://img.cochemist.com/ccimg/50500/50496-15-6.png)
![Ethanone, 1-(3-methoxy[1,1'-biphenyl]-4-yl)-](http://img.cochemist.com/ccimg/50500/50496-15-6_b.png)


![8-Oxabicyclo[3.2.1]octa-2,6-diene, 2,3,4,4-tetrabromo-](http://img.cochemist.com/ccimg/20200/20137-90-0.png)
![8-Oxabicyclo[3.2.1]octa-2,6-diene, 2,3,4,4-tetrabromo-](http://img.cochemist.com/ccimg/20200/20137-90-0_b.png)

![4,13,22,31,37,38,39,40-Octaoxapentacyclo[32.2.1.17,10.116,19.125,28]tetracontane-3,12,21,30-tetrone,2,5,11,14,20,23,29,32-octamethyl-, (1R,2R,5R,7R,10S,11S,14S,16S,19R,20R,23R,25R,28S,29S,32S,34S)-](http://img.cochemist.com/ccimg/6900/6833-84-7.png)
![4,13,22,31,37,38,39,40-Octaoxapentacyclo[32.2.1.17,10.116,19.125,28]tetracontane-3,12,21,30-tetrone,2,5,11,14,20,23,29,32-octamethyl-, (1R,2R,5R,7R,10S,11S,14S,16S,19R,20R,23R,25R,28S,29S,32S,34S)-](http://img.cochemist.com/ccimg/6900/6833-84-7_b.png)




![18-Norandrosta-5,8,11,13-tetraeno[6,5,4-bc]furan-3,7,17-trione,1-hydroxy-2-methoxy-, (1b,2b)-](http://img.cochemist.com/ccimg/3400/3306-52-3.png)
![18-Norandrosta-5,8,11,13-tetraeno[6,5,4-bc]furan-3,7,17-trione,1-hydroxy-2-methoxy-, (1b,2b)-](http://img.cochemist.com/ccimg/3400/3306-52-3_b.png)








![[1,1'-Biphenyl]-4-ol,3-bromo-](http://img.cochemist.com/ccimg/100/92-03-5.png)
![[1,1'-Biphenyl]-4-ol,3-bromo-](http://img.cochemist.com/ccimg/100/92-03-5_b.png)








![8-Oxabicyclo[3.2.1]octa-3,6-dien-2-one, 3-bromo-4-phenyl-](http://img.cochemist.com/ccimg/657400/657389-28-1.png)
![8-Oxabicyclo[3.2.1]octa-3,6-dien-2-one, 3-bromo-4-phenyl-](http://img.cochemist.com/ccimg/657400/657389-28-1_b.png)
![8-Oxabicyclo[3.2.1]octa-3,6-dien-2-one, 3-bromo-4-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/444600/444586-45-2.png)
![8-Oxabicyclo[3.2.1]octa-3,6-dien-2-one, 3-bromo-4-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/444600/444586-45-2_b.png)



![Silane, (1,1-dimethylethyl)dimethyl[(2-methyl-3-furanyl)methoxy]-](http://img.cochemist.com/ccimg/125300/125214-88-2.png)
![Silane, (1,1-dimethylethyl)dimethyl[(2-methyl-3-furanyl)methoxy]-](http://img.cochemist.com/ccimg/125300/125214-88-2_b.png)






![2,3-DIBROMO-8-OXABICYCLO[3.2.1]OCTA-2,6-DIEN-4-ONE](/data/chemimg/65400/444586-28-1.png)
![2,3-DIBROMO-8-OXABICYCLO[3.2.1]OCTA-2,6-DIEN-4-ONE](/data/chemimg/65400/444586-28-1_b.png)

