Co-reporter:Jie Xing, Meitong Zang, Huixiang Liu
Analytica Chimica Acta 2017 Volume 993(Volume 993) pp:
Publication Date(Web):15 November 2017
DOI:10.1016/j.aca.2017.08.047
•Metabolite identification of a drug combination using a novel high resolution mass spectrometry (HRMS)-based technique.•Untargeted background subtraction (BS) followed by a novel ring double bond filtering (RDB) was applied.•In parallel, MDF can be used to recover potential metabolites with similar RDB.•New metabolites were characterized based on HR-MSn data.Metabolite profiling of combination drugs in complex matrix is a big challenge. Development of an effective data mining technique for simultaneously extracting metabolites of one parent drug from both background matrix and combined drug-related signals could be a solution. This study presented a novel high resolution mass spectrometry (HRMS)-based data-mining strategy to fast and comprehensive metabolite identification of combination drugs in human. The model drug combination was verapamil-irbesartan (VER-IRB), which is widely used in clinic to treat hypertension. First, mass defect filter (MDF), as a targeted data mining tool, worked effectively except for those metabolites with similar MDF values. Second, the accurate mass-based background subtraction (BS), as an untargeted data-mining tool, was able to recover all relevant metabolites of VER-IRB from the full-scan MS dataset except for trace metabolites buried in the background noise and/or combined drug-related signals. Third, the novel ring double bond (RDB; valence values of elements in structure) filter, could show rich structural information in more sensitive full-scan MS chromatograms; however, it had a low capability to remove background noise and was difficult to differentiate the metabolites with RDB coverage. Fourth, an integrated strategy, i.e., untargeted BS followed by RDB, was effective for metabolite identification of VER and IRB, which have different RDB values. Majority of matrix signals were firstly removed using BS. Metabolite ions for each parent drug were then isolated from remaining background matrix and combined drug-related signals by imposing of preset RDB values/ranges around the parent drug and selected core substructures. In parallel, MDF was used to recover potential metabolites with similar RDB. As a result, a total of 74 metabolites were found for VER-IRB in human plasma and urine, among which ten metabolites have not been previously reported in human. The results demonstrated that the combination of accurate mass-based multiple data-mining techniques, i.e., untargeted background subtraction followed by ring double bond filtering in parallel with targeted mass defect filtering, can be a valuable tool for rapid metabolite profiling of combination drug.Download high-res image (131KB)Download full-size image
Co-reporter:Tian-Yu Cai, Yun-Rui Zhang, Jian-Bo Ji, Jie Xing
Journal of Ethnopharmacology 2017 Volume 207(Volume 207) pp:
Publication Date(Web):31 July 2017
DOI:10.1016/j.jep.2017.06.025
Ethnopharmacological relevanceThe chemical matrix of the herb Artemisia annua L. (A. annua), from which artemisinin (QHS) is isolated, can enhance both the bioavailability and efficacy of QHS. However, the exact mechanism of this synergism remains unknown. The biotransformation of QHS and potential “enzyme inhibitors” in plant matrix could be of great importance in understanding the improved efficacy of QHS in A. annua, which has been limited to the synergism with flavonoid components.Aim of the studyTo investigate the component in A. annua extracts (MAE) leading to enhanced antiplasmodial potency of QHS via regulation of its metabolism. The efficacy of QHS in combination with the synergistic component was also evaluated.Materials and methodsThe total MAE extract and its three MAE fractions (MAE-I eluted using 3% methanol, MAE-II eluted using 50% methanol and MAE-III eluted using 85% methanol) were obtained from dry plant materials and prepared after lyophilization. The pharmacokinetic profiles of QHS and its major phase I metabolite monohydroxylated artemisinin (QHS-M) were investigated in healthy rats after a single oral administration of QHS in each MAE extract. Major components isolated from the target MAE fraction were evaluated for their enzyme inhibition. The antimalarial activity of QHS in combination with the potential synergistic component against Plasmodium falciparum was studied in vivo (murine Plasmodium yoelii). The recrudescence and survival time of infected mice were also recorded after drug treatment.ResultsCompared to pure QHS, a 2-fold increase in QHS exposure (AUC and Cmax) was found in healthy rats after a single oral dose of QHS in the total MAE extract or its fraction MAE-III. In addition, metabolic biotransformation of QHS to the metabolite QHS-M (mediated by CYP3A) was inhibited by MAE or MAE-III. Among nine major components isolated from MAE-III (five sesquiterpenenes, three flavonoids and one phenolic acid), only arteannuin B (AB) showed an inhibition of CYP3A4 (IC50 1.2 μM). The synergism between QHS and AB was supported using in vivo antiplasmodial assay and a pharmacokinetic study in mice. Unfortunately, the synergism cannot reduce the rate of recrudescence.ConclusionsAB was one of main contributors in A. annua leading to enhanced antiplasmodial potency of QHS via regulation of its metabolism. The final recrudescence indicated the careful use of A. annua for malaria treatment unless additional contributing components or antiplasmodial mechanism were found.Download high-res image (306KB)Download full-size image
Co-reporter:Jie Xing, Meitong Zang, Haiying Zhang, Mingshe Zhu
Analytica Chimica Acta 2015 Volume 897() pp:34-44
Publication Date(Web):15 October 2015
DOI:10.1016/j.aca.2015.09.034
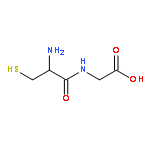
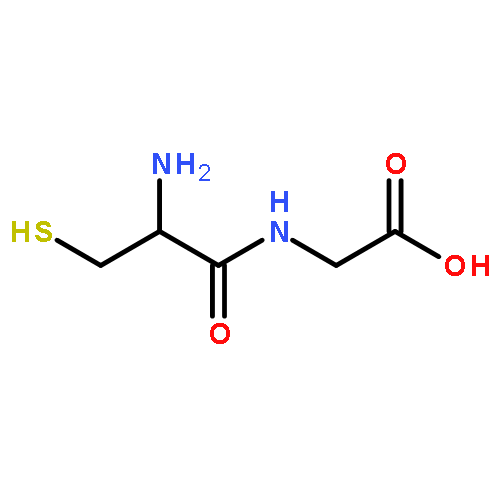
•Metabolite identification of a triple drug combination in humans using high resolution mass spectrometry (HRMS).•Metabolite detection using untargeted background subtraction (BS) followed by improved mass defect filtering (MDF).•Both novel metabolite MDF templates based on BS-processed data and common MDF templates were used.•Unusual metabolites were characterized based on HR-MSn data.Patients are usually exposed to multiple drugs, and metabolite profiling of each drug in complex biological matrices is a big challenge. This study presented a new application of an improved high resolution mass spectrometry (HRMS)-based data-mining tools in tandem to fast and comprehensive metabolite identification of combination drugs in human. The model drug combination was metronidazole-pantoprazole-clarithromycin (MET-PAN-CLAR), which is widely used in clinic to treat ulcers caused by Helicobacter pylori. First, mass defect filter (MDF), as a targeted data processing tool, was able to recover all relevant metabolites of MET-PAN-CLAR in human plasma and urine from the full-scan MS dataset when appropriate MDF templates for each drug were defined. Second, the accurate mass-based background subtraction (BS), as an untargeted data-mining tool, worked effectively except for several trace metabolites, which were buried in the remaining background signals. Third, an integrated strategy, i.e., untargeted BS followed by improved MDF, was effective for metabolite identification of MET-PAN-CLAR. Most metabolites except for trace ones were found in the first step of BS-processed datasets, and the results led to the setup of appropriate metabolite MDF template for the subsequent MDF data processing. Trace metabolites were further recovered by MDF, which used both common MDF templates and the novel metabolite-based MDF templates. As a result, a total of 44 metabolites or related components were found for MET-PAN-CLAR in human plasma and urine using the integrated strategy. New metabolic pathways such as N-glucuronidation of PAN and dehydrogenation of CLAR were found. This study demonstrated that the combination of accurate mass-based multiple data-mining techniques in tandem, i.e., untargeted background subtraction followed by targeted mass defect filtering, can be a valuable tool for rapid metabolite profiling of combination drugs in vivo.
Co-reporter:Xinxiu Li;Peihong Fan;Meitong Zang
Phytochemical Analysis 2015 Volume 26( Issue 1) pp:15-22
Publication Date(Web):
DOI:10.1002/pca.2531
ABSTRACT
Introduction
Soybean protein hydrolysates (SPHs), especially oligopeptides, have shown a variety of functional properties, including immunomodulatory and anti-oxidant effects. Soybean protein hydrolysate products have been used as functional ingredients in food, sports nutrition or clinical nutrition. However, the mixture is mostly undefined due to its complex nature, containing peptides and minor amino acids as well as small proteins.
Objectives
To develop a specific and efficient method for the identification and structural characterisation of oligopeptides in SPHs, and to determine free amino acids in SPHs in the same analytical run, for evaluation of the chemical profile of SPH products.
Methods
Accurate mass spectrometry (MS) datasets of SPH samples were recorded on a high-performance liquid chromatography (HPLC) tandem high-resolution (HR) MS system. Potential oligopeptides were tentatively characterised based on their elemental compositions and ring double bond equivalent (RDBE) values, as well as HRMS/MS data. The analytical method to determine amino acids was evaluated in terms of linearity, precision, apparent recovery and limits of detection and quantitation.
Results
In total, 186 oligopeptides spanning the mass range of m/z 200–1500 and three major free amino acids could be determined in SPH samples in a single sample injection. Ninety-nine oligopeptides were tentatively characterised. The sensitive and specific instrumental performances also permitted the determination of 19 amino acids with a limit of quantitation of ≤ 0.1 μg/mL.
Conclusion
The HPLC–HRMS technique has proven to be an advantageous tool for the rapid characterisation of oligopeptides and determination of amino acids in soybean protein hydrolysates. Copyright © 2014 John Wiley & Sons, Ltd.
Co-reporter:Fuying Du;Zhaohua Liu;Xinxiu Li
Journal of Applied Toxicology 2014 Volume 34( Issue 8) pp:878-884
Publication Date(Web):
DOI:10.1002/jat.2906
ABSTRACT
Triptolide (TP) shows promising anti-inflammatory and antitumor activity but with severe toxicity. TP is a natural reactive electrophile containing three epoxide groups, which are usually linked to hepatotoxicity via their ability to covalently bind to cellular macromolecules. In this study, metabolic pathways leading to detoxification of TP were evaluated in glutathione (GSH)-depleted (treated with L-buthionine-S,R-sulfoxinine, BSO) and aminobenzotriazole (ABT; a non-specific inhibitor for P450s)-treated mice. The toxicity of TP in mice was evaluated in terms of mortality and levels of serum alanine transaminase (ALT). In incubates with NADPH- and GSH-supplemented liver microsomes, seven GSH conjugates derived from TP were detected. In mice, these hydrolytically unstable GSH conjugates underwent γ-glutamyltranspeptidase/dipeptidases-mediated hydrolysis leading to two major cysteinylglycine conjugates, which underwent further hydrolysis by dipeptidases to form two cysteine conjugates of TP. In ABT-treated mice, the hydroxylated metabolites of TP were found at a lower level than normal mice, and their subsequent conjugated metabolites were not found. The level of cysteinylglycine and cysteine conjugates derived from NADPH-independent metabolism increased in mice treated with both TP and BSO (or ABT), which could be the stress response to toxicity of TP. Compared with normal mice, mortality and ALT levels were significantly higher in TP-treated mice, indicating the toxicity of TP. Pretreatment of ABT increased the toxicity caused by TP, whereas the mortality decreased in GSH-depleted mice. Metabolism by cytochrome P450 enzymes to less reactive metabolites implied a high potential for detoxification of TP. The GSH conjugation pathway also contributed to TP's detoxification in mice. Copyright © 2013 John Wiley & Sons, Ltd.
Co-reporter:Meitong Zang, Lixia Zhao, Fanping Zhu, Xinxiu Li, ... Jie Xing
Drug Metabolism and Pharmacokinetics (February 2015) Volume 30(Issue 1) pp:123-126
Publication Date(Web):1 February 2015
DOI:10.1016/j.dmpk.2014.10.008
Repeated pretreatment with the antimalarial drug artemisinin (QHS) could lead to reduced exposure to the parent drug, which is mainly mediated by auto-induction of CYP2B6 activity. CYP2B6 is most sensitive to the inductive effect of constitutive androstane receptor (CAR), which can be activated by QHS. CYP2B6 polymorphism has no influence on pharmacokinetics of QHS derivatives. This study aimed to investigate the effect of CAR (C540T) polymorphism on the auto-induction metabolism-mediated pharmacokinetics of QHS. Healthy Chinese subjects (six in each group with the genotypes of CAR 540C/C, 540C/T and 540T/T; all carrying the CYP2B6*1*1 genotype) received a recommended two-day oral doses of QHS-piperaquine (PQ) to assess the pharmacokinetics of QHS and its metabolite deoxyartemisinin (DQHS). The exposures to QHS and DQHS were significantly lower (p < 0.05) in subjects homozygous for the CAR 540T/T genotype than those with the 540C/C genotype after the repeated dose. QHS did not show different induction clearance in subjects homozygous for the 540C/C genotype (1.3-fold), compared with those carrying the heterozygous 540C/T (2.1-fold) or homozygous 540T/T (1.7-fold) genotype. In conclusion, the CAR (C540T) genotype contributed to the interindividual variability of QHS pharmacokinetics, and the dose regimen for QHS deserves further evaluation especially in specific populations.

![10aH-9,10b-Epoxypyrano[4,3,2-jk][2]benzoxepin-2(3H)-one,octahydro-3,6,9-trimethyl-, (3R,3aS,6R,6aS,9S,10aS,10bR)-](http://img.cochemist.com/ccimg/72900/72826-63-2.png)
![10aH-9,10b-Epoxypyrano[4,3,2-jk][2]benzoxepin-2(3H)-one,octahydro-3,6,9-trimethyl-, (3R,3aS,6R,6aS,9S,10aS,10bR)-](http://img.cochemist.com/ccimg/72900/72826-63-2_b.png)
![8,11-Epoxy-9,12-ethano-11,15-methano-5H,11H-[1,9]dioxacyclooctadecino[4,3-b]pyridine-5,17(18H)-dione,10,13,22,23-tetrakis(acetyloxy)-12-[(acetyloxy)methyl]-14-(benzoyloxy)-7,8,9,10,12,13,14,15,19,20-decahydro-21-hydroxy-8,18,21-trimethyl-,(8R,9R,10R,11S,12S,13R,14R,15S,18S,21S,22S,23R)-](http://img.cochemist.com/ccimg/11100/11088-09-8.png)
![8,11-Epoxy-9,12-ethano-11,15-methano-5H,11H-[1,9]dioxacyclooctadecino[4,3-b]pyridine-5,17(18H)-dione,10,13,22,23-tetrakis(acetyloxy)-12-[(acetyloxy)methyl]-14-(benzoyloxy)-7,8,9,10,12,13,14,15,19,20-decahydro-21-hydroxy-8,18,21-trimethyl-,(8R,9R,10R,11S,12S,13R,14R,15S,18S,21S,22S,23R)-](http://img.cochemist.com/ccimg/11100/11088-09-8_b.png)