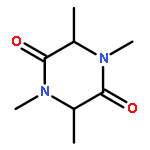
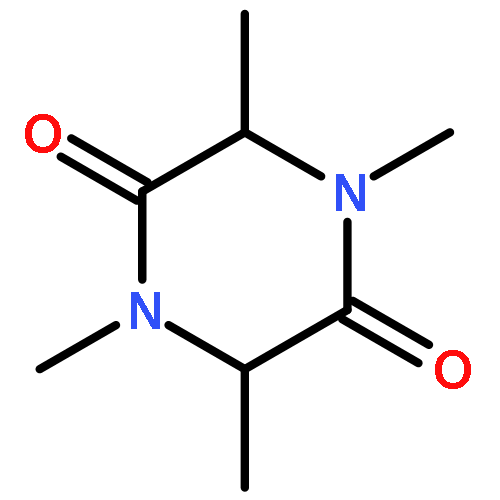
Co-reporter:Haiwei Ni, Yu Zhang, Fang Zhang, Jianying Zhao, Liubi Wu, Xiaozhong Chu
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2015 Volume 138() pp:648-659
Publication Date(Web):5 March 2015
DOI:10.1016/j.saa.2014.11.058
•The vibrational assignments of DNOI have been carried.•Density plots of HOMO–LUMO energy surface plotted to identify donor and acceptor.•Experimentally and computational UV–Vis of the title compound are agree very well.•ESP surface plotted to get electrophilic and nucleophilic sites of the molecule.3-(2,6-Dichlorobenzyl)-5-methyl-N-nitro-1,3,5-oxadiazinan-4-imine (DNOI) was synthesized and characterized by X-ray diffraction, FT-IR, FT-Raman and UV–Vis spectra. The X-ray diffraction study showed that DNOI has a one dimensional configuration, due to the intermolecular C9H⋯O1 and N4H⋯O2 hydrogen bonds. The benzene ring and the oxadiazine rings are tilted with respect to each other by 63.07° (C3N1C5C6). Vibrational spectra and electronic spectra measurements were made for the compound. Optimized geometrical structure and harmonic vibrational frequencies were computed with DFT (B3LYP, B3P86, and M062X) methods using 6-311++G(d,p) basis set. Assignments of the observed spectra were proposed. The equilibrium geometries computed by all of the methods were compared with X-ray diffraction results. The absorption spectra of the title compound were computed both in gas phase and in CH3OH solution using TD-B3LYP/6-311++G(d,p) and PCM-B3LYP/6-311++G(d,p) approaches, respectively. The calculated results provide a good description of positions of the bands maxima in the observed electronic spectrum. Temperature dependence of thermodynamic parameters in the range of 100–1000 K were determined, entropy, heat capacity and enthalpy changes were increasing with temperature increasing, while for Gibbs free energy is decreasing with temperature increasing. The bond orbital occupancies, contribution from parent natural bond orbital (NBO), the natural atomic hybrids was calculated and discussed.In this work, complete vibrational assignments, electronic transitions within molecule and HOMO and LUMO energy of DNOI were studied. MESP map shows that the negative potential sites (red and yellow) are electronegative oxygen atoms, while the positive potential sites (blue) are around the hydrogen atoms. Thermodynamic properties of the title compound were also calculated.
Co-reporter:Yiwei Wang, Yu Zhang, Haiwei Ni, Nana Meng, Kuirong Ma, Jianying Zhao, Dunru Zhu
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2015 Volume 135() pp:296-306
Publication Date(Web):25 January 2015
DOI:10.1016/j.saa.2014.06.103
•The FT-IR, FT-Raman spectra and UV–Vis of the title compound have been recorded experimentally.•Optimized geometry, vibrational frequencies, IR and Raman intensities are obtained with RHF and six DFT methods.•The absorption spectra of the compound were computed both in gas-phase and in DMF solution.•The hybridization and covalent effects in polyatomic wave functions has been studied by analysis the NBO.The compound 9-p-tolyl-9H-carbazole-3-carbaldehyde (HCCD) was synthesized and characterized by X-ray diffraction, FT-IR, FT-Raman and UV–Vis spectra. The X-ray diffraction study showed that HCCD has a Z-configuration. The benzene ring including methyl is twisted from the mean plane of the carbazole group by 59.7(3)°, which is comparable with the calculated result 65° for B3LYP/6-311++G(d, p) method. Vibrational spectra and electronic spectra measurements were made for the compound. Optimized geometrical structure and harmonic vibrational frequencies were computed with B-based DFT (BLYP, B3LYP and cam-B3LYP) methods, and WB-based DFT (WB97, WB97X and WB97XD) methods and ab initio RHF method using 6-311++G(d, p) basis set. Assignments of the observed spectra were proposed. The equilibrium geometries computed by all of the methods were compared with X-ray diffraction results. The absorption spectra of the title compound were computed both in gas phase and in DMF solution using TD-(cam)B3LYP/6-311++G(d, p) and PCM-(cam)B3LYP/6-311++G(d, p) approaches, respectively. The calculated results provide good descriptions of the bands maxima in the observed electronic spectrum. Temperature dependence of thermodynamic parameters in the range of 100–1000 K was determined. The natural atomic hybrids were calculated and discussed.Graphical abstractDFT study of the structural and spectroscopic properties of HCCD has been reported.
Co-reporter:Kang Li, Guodong Tang, ShanShan Kou, Lance F. Culnane, Yu Zhang, Yinglin Song, Rongqing Li, Changmei Wei
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2015 Volume 139() pp:54-62
Publication Date(Web):15 March 2015
DOI:10.1016/j.saa.2014.12.045
•Metal-2-(4,5-diphenyl-1-H-imidazole-2-yl)-phenol complexes have been synthesized.•Structures have been determined by the X-ray diffraction technique.•The geometry and molecular orbitals are calculated based on B3LYP/LANL2DZ.•The third nonlinear optical properties were measured by the Z-scan technique.Three complexes of M(DPIP)2 (M = Cu, Co, Zn as 1, 2, 3) were synthesized and characterized by elemental analysis, IR, UV–Vis, thermogravimetry, and X-ray diffraction. Their nonlinear optical properties were measured by the Z-scan technique and yielded a normalized transmittance of about 70% for complex 1 (45 μJ pulse), and 93% for complex 3 (68 μJ pulse at the focus point). The nonlinear absorption coefficient, β, is 1.4 × 10−11 m/W for 1 and 5.6 × 10−13 m/W for 3, and the third-order nonlinear refraction index, n2, is 1.0 × 10−18 m2/W for 3. Complex 1 shows self-defocusing property, while complex 3 exhibits self-focusing property. The thermogravimetric results show that the frame structure of compounds 1–3 begin to collapse at 400, 250 and 280 °C, respectively, which suggests that they elicit excellent thermal stability. This research aims to provide better understanding of these compounds, and offer preliminary explanations for the significant differences between compounds 1–3, in order to potentially help in the designing of future novel materials with NLO properties.
Co-reporter:Jiahong Wang, Yu Zhang, Zaichao Zhang and Yan Xu
CrystEngComm 2014 vol. 16(Issue 23) pp:5103-5109
Publication Date(Web):31 Mar 2014
DOI:10.1039/C3CE42585A
Three 3-D aluminogermanates L-[C2NH8][AlGe3O8] (1), D-[C2NH8][AlGe3O8] (2) and [Ni(en)3][Al3Ge3O12(H2en)0.5] (3) (en = 1,2-ethanediamine) have been successfully synthesized through a hydrothermal synthesis method (1 and 2 were obtained as a mixture in one autoclave). Crystal structural analysis reveals that three compounds, 1, 2 and 3, are made up of GeO4 (AlO4) tetrahedra. Compounds 1 and 2 with a typical GIS topology are composed of 4-rings, while 3 is constructed exclusively from 3-rings with a JST topology. Compounds 1 and 2 are enantiomers and crystallize in chiral space groups P43212 and P41212, respectively. In compound 3, rigid chiral transition-metal complex cations, [Ni(en)3]2+, and protonated ethylenediamine cations work together as structure directing agents (SDAs) and induce two different kinds of cages ([38·106] and [34·6·103]), which further construct the final structure.
Co-reporter:Fang Zhang, Yu Zhang, Haiwei Ni, Kuirong Ma, Rongqing Li
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2014 Volume 118() pp:162-171
Publication Date(Web):24 January 2014
DOI:10.1016/j.saa.2013.08.076
•The FT-IR, FT-Raman spectra and UV–vis of the title compound have been recorded experimentally.•Optimized geometry, vibrational frequencies are obtained with six DFT methods.•The complete assignments of the experimental spectra are performed on the basis of PED.•The HOMO and LUMO energies have been calculated.•The absorption spectra of the compound were computed both in gas-phase and in H2O solution.Vibrational and electronic spectral measurements were performed for 3-(2-chloro-1,3-thiazol-5-ylmethyl)-5-methyl-1,3,5-oxadiazinan-4-ylidene(nitro) amine (thiamethoxam). Optimized geometrical structure and harmonic vibrational frequencies were calculated with ab initio RHF and DFT (B3LYP, CAMB3LYP, M06 and PBE1PBE) methods with 6-311++G (d, p) basis set. Complete assignments of the observed spectra were proposed. The absorption spectra of the compound were computed in gas-phase using TD-B3LYP/6-311++G (d, p) approach and H2O solution using PCM-TD-B3LYP/6-311++G (d, p) approach. The calculated results matched well with the experimental values. Temperature dependence of thermodynamic parameters in the range of 100–1000 K were determined. The bond orbital occupancies, contribution from parent natural bond orbital (NBO), the natural atomic hybrids was discussed.Graphical abstractDFT study of the structural and spectroscopic properties of thiamethoxam has been reported
Co-reporter:Nana Meng, Yu Zhang, Yiwei Wang, Kuirong Ma, Jianying Zhao, Guodong Tang
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2014 Volume 121() pp:494-507
Publication Date(Web):5 March 2014
DOI:10.1016/j.saa.2013.10.108
•The FT-IR, FT-Raman spectra and UV–Vis of the title compound have been recorded experimentally.•Optimized geometry, vibrational frequencies, IR and Raman intensities are obtained with RHF and six DFT methods.•The complete assignments of the experimental spectra are performed on the basis of the potential energy distribution (PED).•The absorption spectra of the compound were computed both in gas-phase and in DMF solution.2-Amino-3-((E)-(9-p-tolyl-9H-carbazol-3-yl) methyleneamino) maleonitrile (ACMM) was synthesized and characterized by X-ray diffraction, FT-IR, FT-Raman and UV–Vis spectra. The X-ray diffraction study showed that ACMM has a Z-configuration, due to the intramolecular C18H18A⋯N2, N3H3A⋯N2 and C20H20A⋯N4 hydrogen bonds and intermolecular C10H10A⋯N4, N3H3B⋯N9 (2 − x, 2 − y, 2 − z) and N3H8C⋯N4 (2 − x, 1 − y, 2 − z) hydrogen bonds. The benzene ring including methyl is twisted from the mean plane of the carbazole group by 59.7(3)°. Vibrational spectra and electronic spectra measurements were made for the compound. Optimized geometrical structure and harmonic vibrational frequencies were computed with DFT (B3-based B3P86, B3LYP, B3PW91 and B-based BP86, BLYP, BPW91) methods and ab initio RHF method using 6-311++G(d, p) basis set. Assignments of the observed spectra were proposed. The equilibrium geometries computed by all of the methods were compared with X-ray diffraction results. The absorption spectra of the title compound were computed both in gas phase and in DMF solution using TD-B3LYP/6-311++G(d, p) and PCM-B3LYP/6-311++G(d, p) approaches, respectively. The calculated results provide a good description of positions of the bands maxima in the observed electronic spectrum. Temperature dependence of thermodynamic parameters in the range of 100–1000 K were determined. The bond orbital occupancies, contribution from parent natural bond orbital (NBO), the natural atomic hybrids was calculated and discussed.Graphical abstractDFT study of the structural and spectroscopic properties of ACMM has been reported.
Co-reporter:Jian-Ying Zhao, Yu Zhang, Feng-Qi Zhao, and Xue-Hai Ju
The Journal of Physical Chemistry A 2013 Volume 117(Issue 47) pp:12519-12528
Publication Date(Web):November 7, 2013
DOI:10.1021/jp405934w
The adsorption of a CO2 molecule on neutral and charged X-centered icosahedron Al12X±z clusters (X = Al, Be, Zn, Ni, Cu, B, P; z = 0, 1) was investigated by the density functional PW91 and PWC methods. Optimized configurations corresponding to physisorption and chemisorption of CO2 were identified. The adsorption energies, activation barriers, and binding energies involving both the physisorption (Al12X±z·CO2–I) and chemisorption (Al12X±z·CO2–II) for CO2 were determined. The chemisorption of a CO2 molecule on the Al12X clusters (X is a metallic doping element) requires relatively low activation barriers. The lowest barrier was found to be with the Al12Be cluster. For the Al12X– clusters, the barriers are all higher than those of the neutral analogues. For the Al12X+ clusters, two corresponding configurations are linked by a low-energy barrier, and CO2 molecule chemisorption on the Al12Be+ cluster has the lowest barrier. The adsorption energies are larger than the energy barriers, which facilitates the chemisorption. The results show that carbon dioxide adsorbed on the Al12X±z clusters can be tuned by controllable X doping and the total number of valence electrons and suggest the potential application of Al12X±z nanostructures for carbon dioxide capture and activation.
Co-reporter:Yu Zhang, Fang Zhang, Kuirong Ma, Guodong Tang
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2013 Volume 105() pp:352-358
Publication Date(Web):15 March 2013
DOI:10.1016/j.saa.2012.11.063
Vibrational and electronic spectral measurements were made for 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ). Optimized geometrical structure and harmonic vibrational frequencies were computed by DFT(B3LYP, CAMB3LYP, B3P86, M062X, mPW3PBE and PBE1PBE) using 6-311++G(d,p) basis set. Complete assignments of the observed spectra were proposed. The absorption spectra of the compound were computed both in gas-phase and in CH2Cl2 solution using TDCAMB3LYP/6-311++G(d,p) and PCM-TDCAMB3LYP/6-311++G(d,p) approaches, respectively, the calculated results provide a good description of positions of the bands maxima in the observed electronic spectrum.Graphical abstractDFT study of the structural and spectroscopic properties of DDQ has been reported.Highlights► The FT-IR, FT-Raman spectra and UV–vis of the title compound have been recorded experimentally. ► Optimized geometry, vibrational frequencies, IR and Raman intensities are obtained with six DFT methods. ► The complete assignments of the experimental spectra are performed on the basis of the potential energy distribution (PED). ► The HOMO and LUMO energies have been calculated. ► The absorption spectra of the compound were computed both in gas-phase and in CH2Cl2 solution.
Co-reporter:Hu Zhou, Yu Zhang, Dun-ru Zhu
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2012 Volume 86() pp:20-26
Publication Date(Web):February 2012
DOI:10.1016/j.saa.2011.09.034
The structures and some molecular properties of the complexes M(mnt)22− (M = Ni, Pd, Pt and Zn, Cd, Hg; mnt2− = deprotonated maleonitriledithiolate) have been studied by using density functional theory (DFT) B3LYP/LanL2DZ level of theory. Computed binding energies show that the sequences of binding strengths are Ni < Pd < Pt and Cd < Hg < Zn. The natural bonding orbitals analyses show that Ni, Pd and Pt gain 1.40e, 1.62e and 1.72e, respectively to their n s, (n−1)dx2−y2(n−1)dx2−y2 and (n−1)dz2(n−1)dz2 orbitals from both ligand mnt2− and (n − 1)dyz of metal ions, while Zn, Cd and Hg complexes gain electrons to their ns orbitals. The absorption spectra of these complexes were obtained by using time-dependent density functional theory associated with polarized continuum model. Comparison of the absorption spectra in acetonitrile solution with those in gas phase show that the solvatochromic effect made the lowest energy absorption red shift by 31, 34 and 44 nm for d8 metal complexes Ni(mnt)22−, Pd(mnt)22− and Pt(mnt)22−, respectively, while blue shift by 28, 44, 25 nm for d10 metal complexes Zn(mnt)22−, Cd(mnt)22− and Hg(mnt)22−, respectively. The calculated results reproduced the experimental data with the deviations less than 5% for Ni–S stretching vibrational frequencies and less than 3% for other vibrational modes.Graphical abstractDFT study of the structural and spectroscopic properties of complexes M(mnt)22− (M = Ni, Pd, Pt and Zn, Cd, Hg) has been reported.Highlights► Computed binding energies show that the sequences of binding strengths are Ni(mnt)22− < Pd(mnt)22− < Pt(mnt)22− and Cd(mnt)22− < Hg(mnt)22− < Zn(mnt)22−. ► Comparison of the absorption spectra in acetonitrile solution with those in gas phase show that the solvatochromic effect made the lowest energy absorption red shift. ► The calculated results reproduced the the experimental data with the deviations less than 5% for Ni-S stretching vibrational frequencies and less than 3% for other vibrational modes.
Co-reporter:Yu Zhang, Hu Zhou, Zhengjing Jiang, Rongqing Li
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2011 Volume 83(Issue 1) pp:112-119
Publication Date(Web):December 2011
DOI:10.1016/j.saa.2011.07.091
Vibrational and electronic spectral measurements were made for 4,5-dihydro-6-methyl-4-[(E)-(3-pyridinylmethylene)amino]-1,2,4-triazin-3(2H)-one (pymetrozine). Optimized geometrical structure and harmonic vibrational frequencies were computed by ab initio RHF, B-based DFT methods (BLYP, BP86 and BPW91) and B3-based DFT methods (B3LYP, B3P86 and B3PW91) using 6-311++G(d,p) basis set. Complete assignments of the observed spectra were proposed. The absorption spectra of the compound were computed both in gas-phase and in C2H5OH solution using TD-B3LYP/6-311++G(d,p) and PCM-B3LYP/6-311++G(d,p) approaches, respectively, the calculated results provide a good description of positions of the bands maxima in the observed electronic spectrum. The MEP calculation indicates that the most possible site for electrophilic attack is H23 and the most possible sites for nucleophilic attack are N5 and O19.Graphical abstractThe Fourier-transform infrared and Raman spectra and UV–vis spectra of 4,5-dihydro-6-methyl-4-[(E)-(3-pyridinylmethylene)amino]-1,2,4-triazin-3(2H)-one (pymetrozine) have been recorded experimently and analyzed with the aid of B-based (BLYP, BP86 and BPW91) and B3-based (B3LYP, B3P86 and B3PW91) DFT methods.Highlights► For 4,5-dihydro-6-methyl-4-[(E)-(3-pyridinylmethylene)amino]-1,2,4-triazin-3(2H)-one, the observed frequencies are reproduced well by the B-based DFT methods. ► The calculated band maximums at 219.8, 245.7 and 298.5 nm in gas phase (217.6, 246.4 and 313.4 nm in C2H5OH solution, respectively) provide a good description on positions of the band maximums in the observed electronic spectrum. ► The molecular electrostatic potential (MEP) predict the possible site of pymetrozine for electrophilic attack is H23 and the most possible sites for nucleophilic attack are N5 and O19.
Co-reporter:Yu Zhang, Rongqing Li, Zhengjing Jiang
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2010 Volume 77(Issue 2) pp:378-387
Publication Date(Web):1 October 2010
DOI:10.1016/j.saa.2010.05.018
The vibrational spectra of aluminum halides, AlX3 (X = F, Cl, Br and I) and their dimers, Al2X6, have been systematically investigated by ab initio restricted Hartree–Fock (RHF) and density functional B3LYP and B3P86 methods with LanL2DZ, SDD, CEP-31G and DGDZVP basis sets. The optimized geometries, calculated vibrational frequencies were evaluated via comparing with the experimental data. The vibrational frequencies, calculated by these methods with different basis sets, were compared to each other. The effect of the methods and the basis sets used on the calculated vibrational frequencies was discussed. The best fittings values between the calculated and the measured vibrational frequencies were achieved by B3LYP/DGDZVP theoretical level, with this method, the deviations are less than 2% for Al–X stretching vibrational modes in AlX3 and less than 4% for Al–X stretching vibrational modes in Al2X6. Some vibrational frequencies of Al(III) halides were predicted.
Co-reporter:Yu Zhang, Jianying Zhao, Rongqing Li, Zhengjing Jiang, Guodong Tang
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2010 Volume 77(Issue 4) pp:732-739
Publication Date(Web):November 2010
DOI:10.1016/j.saa.2010.07.004
Vibrational spectral measurements were made for 1,5-dimethyl-2-phenyl-4-[(pyridin-4-ylmethylene)-amino]-1,2-dihydro-pyrazol-3-one (DPPDP). Optimized geometrical structure and harmonic vibrational frequencies were computed by ab initio RHF and DFT (B-based BP86, BLYP, BPW91, B3-based B3P86, B3LYP, B3PW91 and O3-based O3LYP) methods using 6-311++G(d,p) basis set. Complete assignments of the observed spectra were proposed. The equilibrium geometries computed by all of the methods, were compared with X-ray diffraction results. The absorption spectra of the title compound were computed both in gas phase and in CH3CN solution using TD-B3LYP/6-311++G(d,p) and PCM-B3LYP/6-311++G(d,p) approaches and the calculated results provide a good description of positions of the two band maxima in the observed electronic spectrum.
Co-reporter:Guo-dong Tang, Zheng-jing Jiang, Rong-qing Li, Jin-fang Zhang, Yu Zhang, Chi Zhang
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2009 Volume 74(Issue 1) pp:228-232
Publication Date(Web):15 September 2009
DOI:10.1016/j.saa.2009.06.035
Theoretical calculations were carried out on some neutral nest-shaped heterothiometallic cluster compounds [MOS3Py5Cu3X] (M = Mo, W; X = F, Cl, Br, I) with the high first static hyperpolarizabilities β values. The geometries of these cluster compounds were optimized by the restricted DFT method at B3LYP level with LanL2DZ base set without any constrains. In order to understand the relationship between the first static hyperpolarizabilities and the compositions of these clusters, the frontier orbital compositions and energy gaps between the HOMO and LUMO orbitals were calculated and analysed. In these clusters the HOMO orbitals are mainly composed of halogen atoms and the first static hyperpolarizability increases from F to I atom. The LUMO orbitals of clusters [MoOS3Py5Cu3X] are comprised of Mo, O and S atoms while the LUMO orbitals of clusters [WOS3Py5Cu3X] composed of W atom and pyridine ring. The energy gaps between the HOMO and LUMO orbitals of the clusters [MoOS3Py5Cu3X] are smaller than those of the clusters [WOS3Py5Cu3X]. As a result the first static hyperpolarizability values of the clusters [MoOS3Py5Cu3X] are higher than those of the clusters [WOS3Py5Cu3X].