Co-reporter:Cláudio M. Nunes, Stephanie N. Knezz, Igor Reva, Rui Fausto, and Robert J. McMahon
Journal of the American Chemical Society 2016 Volume 138(Issue 47) pp:15287-15290
Publication Date(Web):September 29, 2016
DOI:10.1021/jacs.6b07368
Triplet 2-formyl phenylnitrene was generated by photolysis of 2-formyl phenylazide isolated in Ar, Kr, and Xe matrixes and characterized by IR, UV–vis, and EPR spectroscopies. Upon generation at 10 K, the triplet nitrene spontaneously rearranges in the dark to singlet 6-imino-2,4-cyclohexadien-1-ketene on the time scale of several hours. The intramolecular [1,4] H atom shift from the nitrene to the imino ketene occurs by tunneling, on the triplet manifold, followed by intersystem crossing. This case constitutes the first direct evidence of a tunneling reaction involving a nitrene.
Co-reporter:Stephanie N. Knezz, Terese A. Waltz, Benjamin C. Haenni, Nicola J. Burrmann, and Robert J. McMahon
Journal of the American Chemical Society 2016 Volume 138(Issue 38) pp:12596-12604
Publication Date(Web):September 17, 2016
DOI:10.1021/jacs.6b07444
Photolysis (λ > 472 nm) of 2-diazo-3-pentyne (11) affords triplet 1,3-dimethylpropynylidene (MeC3Me, 33), which was characterized spectroscopically in cryogenic matrices. The infrared, electronic absorption, and electron paramagnetic resonance spectra of MeC3Me (33) are compared with those of the parent system (HC3H) to ascertain the effect of alkyl substituents on delocalized carbon chains of this type. Quantum chemical calculations (CCSD(T)/ANO1) predict an unsymmetrical equilibrium structure for triplet MeC3Me (33), but they also reveal a very shallow potential energy surface. The experimental IR spectrum of triplet MeC3Me (33) is best interpreted in terms of a quasilinear, axially symmetric structure. EPR spectra yield zero-field splitting parameters that are typical for triplet carbenes with axial symmetry (|D/hc| = 0.63 cm–1, |E/hc| = ∼ 0 cm–1), while theoretical analysis suggests that the methyl substituents confer significant spin polarization to the carbon chain. Upon irradiation into the near-UV electronic absorption (λmax 350 nm), MeC3Me (33) undergoes 1,2-hydrogen migration to yield pent-1-en-3-yne (4), a photochemical reaction that is typical of carbenes bearing a methyl substituent. This facile process apparently precludes photoisomerization to other interesting C5H6 isomers, in contrast to the rich photochemistry of the parent C3H2 system.
Co-reporter:Vanessa L. Orr, Brian J. Esselman, P. Matisha Dorman, Brent K. Amberger, Ilia A. Guzei, R. Claude Woods, and Robert J. McMahon
The Journal of Physical Chemistry A 2016 Volume 120(Issue 39) pp:7753-7763
Publication Date(Web):September 27, 2016
DOI:10.1021/acs.jpca.6b07610
The pure rotational spectrum of diketene has been studied in the millimeter-wave region from ∼240 to 360 GHz. For the ground vibrational state and five vibrationally excited satellites (ν24, 2ν24, 3ν24, 4ν24, and ν16), the observed spectrum allowed for the measurement, assignment, and least-squares fitting a total of more than 10 000 distinct rotational transitions. In each case, the transitions were fit to single-state, complete or near-complete sextic centrifugally distorted rotor models to near experimental error limits using Kisiel’s ASFIT. Additionally, we obtained less satisfactory least-squares fits to single-state centrifugally distorted rotor models for three additional vibrational states: ν24 + ν16, ν23, and 5ν24. The structure of diketene was optimized at the CCSD(T)/ANO1 level, and the vibration–rotation interaction (αi) values for each normal mode were determined with a CCSD(T)/ANO1 VPT2 anharmonic frequency calculation. These αi values were helpful in identifying the previously unreported ν16 and ν23 fundamental states. We obtained a single-crystal X-ray structure of diketene at −173 °C. The bond distances are increased in precision by more than an order of magnitude compared to those in the 1958 X-ray crystal structure. The improved accuracy of the crystal structure geometry resolves the discrepancy between previous computational and experimental structures. The rotational transition frequencies provided herein should be useful for a millimeter-wave or terahertz search for diketene in the interstellar medium.
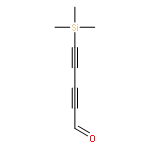
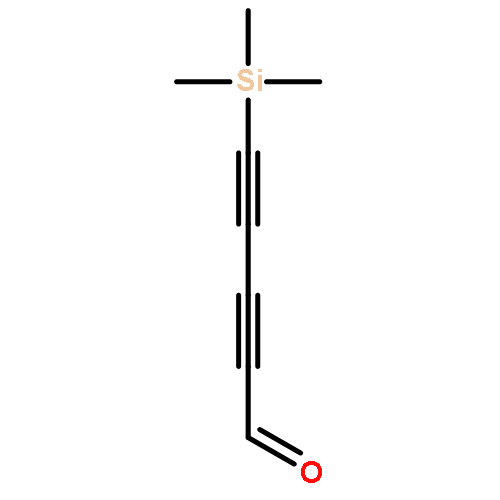
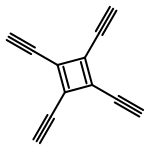
Co-reporter:Brian J. Esselman, Frank L. Emmert III, Andrew J. Wiederhold, Stephanie J. Thompson, Lyudmila V. Slipchenko, and Robert J. McMahon
The Journal of Organic Chemistry 2015 Volume 80(Issue 23) pp:11863-11868
Publication Date(Web):October 28, 2015
DOI:10.1021/acs.joc.5b01864
The mechanism by which carbon condenses to form PAHs or fullerenes is a problem that has garnered considerable theoretical and experimental attention. The ring-coalescence and annealing model for the formation of C60 involves a [2 + 2] cycloaddition reaction of a cyclopolyyne to form a tetraalkynyl cyclobuta-1,3-diene intermediate, followed by a Bergman cycloaromatization reaction of the enediyne moiety. Intramolecular trapping of the incipient p-benzyne diradical across a diyne moiety of the macrocyclic ring affords an aromatic ring that must undergo further intramolecular reactions via polyradical intermediates to produce a condensed graphitic structure or fullerene. Computational studies of a model system for the intriguing tetraalkynylcyclobuta-1,3-diene intermediate, however, reveal that the corresponding p-benzyne diradical lies in a shallow minimum with a very low barrier to ring opening to cyclooctadienediyne. This pathway has not been previously considered in the mechanism for carbon condensation.
Co-reporter:Brent K. Amberger, Brian J. Esselman, R. Claude Woods, Robert J. McMahon
Journal of Molecular Spectroscopy 2014 Volume 295() pp:15-20
Publication Date(Web):January 2014
DOI:10.1016/j.jms.2013.11.001
•Carbonyl diazide was synthesized and its millimeter-wave spectrum was collected.•Transitions for two conformations of carbonyl diazide were analyzed.•Transitions for four excited states of the syn–syn form were measured and fit.•The experimental energy separation of conformers matches theoretical predictions.•Distortion and vibration–rotation constants were generally well predicted by theory.Millimeter-wave absorption spectra for carbonyl diazide (OC(N3)2) are reported in the frequency range of 243–360 GHz, at both 293 K and 213 K. Transitions for two of the three possible conformations, one with both of the azide groups syn to the carbonyl group, or with one syn and the other anti, were observed in the spectra. Theoretical calculations at the CCSD(T)/ANO1 level do an excellent job of predicting the ground state rotational constants and 4th order centrifugal distortion terms for both conformers. Relative line intensities, along with theoretically predicted dipole moments, were used to estimate the energy difference of the two observed forms, yielding a result in good agreement with the ab initio potential energy surface. The spectra of the ν12, ν7, ν9 and 2ν12 excited vibrational states for the more abundant syn–syn conformer have been assigned, and a great many transitions for each of them have been fit using partial 6th and 8th order centrifugal distortion Hamiltonians. Anharmonic vibration–rotation interaction constants from the CCSD(T)/ANO1 calculations are in excellent agreement with the experimentally determined constants in the case of ν7 and ν9, but not for ν12.Graphical abstract
Co-reporter:Hiroshi Inui ; Kazuhiro Sawada ; Shigero Oishi ; Kiminori Ushida
Journal of the American Chemical Society 2013 Volume 135(Issue 28) pp:10246-10249
Publication Date(Web):June 24, 2013
DOI:10.1021/ja404172s
In the photodecompositions of 4-methoxyphenyl azide (1) and 4-methylthiophenyl azide (5) in argon matrixes at cryogenic temperatures, benzazirine intermediates were identified on the basis of IR spectra. As expected, the benzazirines photochemically rearranged to the corresponding ketenimines and triplet nitrenes. Interestingly, with the methylthio substituent, the rearrangement of benzazirine 8 to ketenimine 7 occurred at 1.49 × 10–5 s–1 even in the dark at 10 K, despite a computed activation barrier of 3.4 kcal mol–1. Because this rate is 1057 times higher than that calculated for passing over the barrier and because it shows no temperature dependence, the rearrangement mechanism is interpreted in terms of heavy-atom tunneling.
Co-reporter:Caroline R. Pharr ; Laura A. Kopff ; Brian Bennett ; Scott A. Reid
Journal of the American Chemical Society 2012 Volume 134(Issue 14) pp:6443-6454
Publication Date(Web):March 30, 2012
DOI:10.1021/ja300927d
Photolysis (λ > 543 nm) of 3-thienyldiazomethane (1), matrix isolated in Ar or N2 at 10 K, yields triplet 3-thienylcarbene (13) and α-thial-methylenecyclopropene (9). Carbene 13 was characterized by IR, UV/vis, and EPR spectroscopy. The conformational isomers of 3-thienylcarbene (s-E and s-Z) exhibit an unusually large difference in zero-field splitting parameters in the triplet EPR spectrum (|D/hc| = 0.508 cm–1, |E/hc| = 0.0554 cm–1; |D/hc| = 0.579 cm–1, |E/hc| = 0.0315 cm–1). Natural Bond Orbital (NBO) calculations reveal substantially differing spin densities in the 3-thienyl ring at the positions adjacent to the carbene center, which is one factor contributing to the large difference in D values. NBO calculations also reveal a stabilizing interaction between the sp orbital of the carbene carbon in the s-Z rotamer of 13 and the antibonding σ orbital between sulfur and the neighboring carbon—an interaction that is not observed in the s-E rotamer of 13. In contrast to the EPR spectra, the electronic absorption spectra of the rotamers of triplet 3-thienylcarbene (13) are indistinguishable under our experimental conditions. The carbene exhibits a weak electronic absorption in the visible spectrum (λmax = 467 nm) that is characteristic of triplet arylcarbenes. Although studies of 2-thienyldiazomethane (2), 3-furyldiazomethane (3), or 2-furyldiazomethane (4) provided further insight into the photochemical interconversions among C5H4S or C5H4O isomers, these studies did not lead to the spectroscopic detection of the corresponding triplet carbenes (2-thienylcarbene (11), 3-furylcarbene (23), or 2-furylcarbene (22), respectively).
Co-reporter:Alex M. Nolan, Brent K. Amberger, Brian J. Esselman, Venkatesan S. Thimmakondu, John F. Stanton, R. Claude Woods, and Robert J. McMahon
Inorganic Chemistry 2012 Volume 51(Issue 18) pp:9846-9851
Publication Date(Web):August 28, 2012
DOI:10.1021/ic301270b
Carbonyl diazide (1), OC(N3)2, is prepared by reaction of triphosgene and tetra-n-butylammonium azide in a solution of diethyl ether or dimethyl ether. The advantage of this synthetic method, relative to other procedures, is that the use of triphosgene, OC(OCCl3)2, mitigates the need to use highly poisonous reagents such as phosgene, OCCl2, or chlorofluorocarbonyl, OC(Cl)F. The identity and purity of OC(N3)2 are established by gas-phase IR spectroscopy, which reveals the presence of both syn–syn and anti–syn conformers. Computed anharmonic vibrational frequencies and infrared intensities of carbonyl diazide (1) display excellent agreement with experiment, and reveal the presence of strong Fermi resonances.
Co-reporter:Brian J. Esselman and Robert J. McMahon
The Journal of Physical Chemistry A 2012 Volume 116(Issue 1) pp:483-490
Publication Date(Web):December 20, 2011
DOI:10.1021/jp206478q
The effects of ethynyl substitution on cyclobutadiene are explored via density functional theory and coupled-cluster calculations. The computed singlet–triplet gaps indicate a monotonic dependence on the degree of ethynyl substitution, which differentially stabilizes the triplet relative to the singlet ground state and reduces the gap. A series of isodesmic, homodesmotic, and hyperhomodesmotic equations are employed to quantify the stabilization upon ethynyl substitution. Analyses that rely on a simple isodesmic equation and/or B3LYP/6-31G(d) values are found to be problematic. Analyses that rely on homodesmotic or hyperhomodesmotic equations, in conjunction with CCSD/cc-pVDZ values, are more robust. Using a hyperhomodesmotic equation to assess the stabilization enthalpies of tetra-substituted singlet cyclobutadienes, our analysis predicts tetramethylcyclobutadiene (ΔH0rxn = −17.3 kcal/mol) to be more stable than tetraethynylcyclobutadiene (ΔH0rxn = −11.7 kcal/mol), which, in turn, is substantially more stable than tetracyanocyclobutadiene (ΔH0rxn = +12.7 kcal/mol).
Co-reporter:Carl R. Kemnitz, Eric S. Ball, and Robert J. McMahon
Organometallics 2012 Volume 31(Issue 1) pp:70-84
Publication Date(Web):December 12, 2011
DOI:10.1021/om200555e
We report novel photochemistry derived from (η5-C5H5)Mn(CO)3 (1a), (η5-C5H4Me)Mn(CO)3 (1b),(η5-C5Me5)Mn(CO)3 (1c), and (η5-indenyl)Mn(CO)3 (1d). Photolysis (>261 nm, 1 h) of the parent tricarbonyl (1a–d), matrix isolated in argon at 10 K, yields two species: the expected singlet dicarbonyl 1(η5-L)Mn(CO)2 (12a–d) and an additional compound assigned as the triplet dicarbonyl 3(η5-L)Mn(CO)2 (32a–d). Density functional theory calculations (B3LYP/LANL2DZ) support the structural assignments for 12 and 32. Natural bond orbital population analyses of 12a and 32a explain the source of the large coupling (ΔνCO 153 cm–1) between the carbonyl stretching vibrations in 32a. The triplet isomer (32) is metastable, even at temperatures as low as 10 K. We determined the rate constants for the thermal isomerization 32 → 12 using dispersive kinetic analysis. As revealed by these rate constants, the triplet complexes display the following order of stability in this system: Ind ≫ Cp ≈ Cp′ > Cp*. The spectroscopy and kinetics observed in various matrices (Ar, CH4, and Xe) do not differ appreciably. Experimental and computational results suggest that the singlet–triplet energy gap (ΔEST) of CpMn(CO)2 (2a) must be smaller than previous estimates.
Co-reporter:Jessica L. Menke, Eric V. Patterson and Robert J. McMahon
The Journal of Physical Chemistry A 2010 Volume 114(Issue 22) pp:6431-6437
Publication Date(Web):May 13, 2010
DOI:10.1021/jp101963p
The effects of cyano substitution on cyclobutadiene are explored using density functional, coupled-cluster, CASSCF, and CASPT2 calculations. An isodesmic reaction is employed to gauge the relative stabilization (ΔHrxn°) of cyclobutadienes with varying numbers of cyano groups. Although density functional theory predicts a relatively large stabilization for the addition of four cyano substituents to cyclobutadiene (18.5 kcal/mol), coupled-cluster theory predicts a smaller stabilization (9.3 kcal/mol). The effect of the number of cyano groups on the singlet−triplet gaps is also investigated. NBO calculations lend insight into the structural trends of the triplets, and the comparison of coupled-cluster and CASSCF calculations sheds light on the multireference electronic character in these systems. The effect of tetracyano substitution on tetrahedrane and other C4H4 isomers is also explored.
Co-reporter:Dr. Henning Hopf;Cornelia Mlynek;Dr. Robert J. McMahon;Jessica L. Menke;Dr. Alberto Lesarri;Dr. Michael Rosemeyer;Dr. Jens-Uwe Grabow
Chemistry - A European Journal 2010 Volume 16( Issue 47) pp:14115-14123
Publication Date(Web):
DOI:10.1002/chem.201001648
Abstract
In support of a deeper understanding of the chemistry of cyanoacetylene—a known constituent of planetary atmospheres and interstellar space—theoretical and experimental studies address the chemical mechanism of dimerization and trimerization, and provide high-resolution rotational spectra of two of the trimeric products, 1,2,3- and 1,2,4-tricyanobenzene. Analysis of the rotational spectra is particularly challenging because of quadrupolar coupling from three 14N nuclei. The laboratory rotational spectra provide the basis for future searches for these polar aromatic compounds in interstellar space by radio astronomy.
Co-reporter:Christopher J. Shaffer, Brian J. Esselman, Robert J. McMahon, John F. Stanton and R. Claude Woods
The Journal of Organic Chemistry 2010 Volume 75(Issue 6) pp:1815-1821
Publication Date(Web):February 12, 2010
DOI:10.1021/jo9026462
Stimulated and intrigued by the report of the synthesis of diazirinone (1), a metastable adduct of N2 and CO, we carried out further experimental and theoretical studies aimed at the detailed spectroscopic characterization of this species. Our attempts to generate and detect diazirinone (1) in either the condensed phase (using matrix isolation spectroscopy) or in the gas phase (using millimeter-wave rotational spectroscopy), however, have been unsuccessful. Trapping the volatile products produced from the reaction of 3-chloro-3-(p-nitrophenoxy)diazirine (5) with tetrabutylammonium fluoride (TBAF) under matrix-isolation conditions affords chlorofluorodiazirine (8) and carbon monoxide but fails to provide evidence for diazirinone (1). Moreover, sophisticated ab initio calculations of the structure and fundamental vibrational frequencies of diazirinone (1) produce an estimate for the fundamental band origin of the C═O stretch (2046 cm−1) that is ca. 100 cm−1 lower in frequency than the experimental value previously attributed to this band. This discrepancy lies well outside any expected solvent shift or calculation error at this level of theory. In an effort to reconcile our findings with the earlier reports concerning diazirinone (1), we reconsidered the infrared spectral evidence upon which the original claim of diazirinone synthesis was based. New experiments demonstrate that these spectra may be explained and reproduced with a combination of solution-phase and gas-phase absorptions of CO, without recourse to invoke diazirinone (1).
Co-reporter:Phillip S. Thomas ; Nathan P. Bowling
Journal of the American Chemical Society 2009 Volume 131(Issue 24) pp:8649-8659
Publication Date(Web):May 21, 2009
DOI:10.1021/ja901977s
Triplet carbene methylpentadiynylidene, MeC5H (1), was investigated in cryogenic matrices by IR, UV/vis, and EPR spectroscopy. Broadband irradiation (λ > 497 nm) of the isomeric diazo compounds, 1-diazo-hexa-2,4-diyne (2) or 2-diazo-hexa-3,5-diyne (3), generates triplet carbene 1. EPR spectra yield zero-field splitting parameters (|D/hc| = 0.62 cm−1, |E/hc| < 0.0006 cm−1), which are typical for a triplet carbene with axial symmetry. The electronic spectrum of triplet 1 is characterized by a weak absorption in the near-UV and visible region (350−430 nm) with vibronic progressions corresponding to excitations of the acetylenic stretching and the terminal C≡C−H bending modes. Chemical trapping of triplet 1 in an O2-doped matrix affords carbonyl oxides derived predominantly from attack at C-3. Upon irradiation at λ > 399 nm, triplet 1 undergoes photochemical 1,2-hydrogen migration to form hex-1-ene-3,5-diyne (6).
Co-reporter:Randal A. Seburg ; Eric V. Patterson
Journal of the American Chemical Society 2009 Volume 131(Issue 26) pp:9442-9455
Publication Date(Web):June 16, 2009
DOI:10.1021/ja901606a
Spectroscopic data for triplet isotopomers H−C−C−C−H, H−13C−C−C−H, and H−C−13C−C−H are consistent with computational predictions for a symmetric structure in which the terminal carbons are equivalent (C2 or C2v) and are inconsistent with a planar (Cs) structure in which they are not. Experimentally observed 13C isotope shifts in the IR spectra and 13C hyperfine coupling constants in the EPR spectra exhibit good agreement with values predicted by theory for a C2 structure. The 13C hyperfine coupling constants also provide an independent experimental estimate for the bond angles in the molecule. The isotope-dependence of the zero-field splitting parameters reveals the influence of molecular motion in modulating the values of these parameters. The interpretation of motional effects provides a basis for rationalizing the anomalously low E value, which had previously been interpreted in terms of an axially symmetric (D∞h) structure. Computational studies involving Natural Bond Orbital and Natural Resonance Theory analyses provide insight into the spin densities and the complex electronic structure of this reactive intermediate.
Co-reporter:RalA. Seburg;JonathanA. Hodges;RobertJ. McMahon
Helvetica Chimica Acta 2009 Volume 92( Issue 8) pp:1626-1643
Publication Date(Web):
DOI:10.1002/hlca.200800446
Abstract
Mechanistic and spectroscopic investigations of reactive C3H2 hydrocarbons necessitated the preparation of diazopropyne isotopomers bearing mono-13C substitution at each of the three unique positions. The diazo compounds and their tosylhydrazone precursors were prepared from the mono-13C isotopomers of propynal (in the form of either the aldehyde or the diethyl acetal). The introduction of 13C-labeling at either alkyne position in propynal utilized the Corey–Fuchs procedure for chain homologation.
Co-reporter:Nicholas A. Walters, Brent K. Amberger, Brian J. Esselman, R. Claude Woods, Robert J. McMahon
Journal of Molecular Spectroscopy (January 2017) Volume 331() pp:
Publication Date(Web):January 2017
DOI:10.1016/j.jms.2016.11.011
•Rotational spectra for formyl azide (HC(O)N3) were obtained from 240 to 360 GHz.•1573 rotational transitions were observed for the ground state of syn formyl azide.•Six vibrational satellites of syn formyl azide were observed.•Excellent agreement: measured, computed vibration-rotation interaction.•Thermal decomposition kinetics of formyl azide monitored (half-life t½ = 30 min).Millimeter-wave spectra for formyl azide (HC(O)N3) were obtained from 240 to 360 GHz at ambient temperature. For the ground state of syn formyl azide, over 1500 independent rotational transitions were measured and least-squares fit to a complete S-reduced 8th order centrifugal distortion/rigid rotor Hamiltonian. The decomposition of formyl azide was monitored over a period of several hours, the half-life (t½ = 30 min) was determined, and its decomposition products were investigated. Transitions from five vibrational satellites of syn formyl azide (ν9, ν12, 2ν9, ν9 + ν12, and ν11) were observed, measured, and least-squares fit to complete or nearly complete octic centrifugally-distorted, single-state S-reduced models. A less complete single-state fit of 3ν9 (509.3 cm−1) was obtained from an unperturbed subset of its assignable transitions. This state is apparently coupled to the fundamental ν8 (489.4 cm−1) and the overtone 2ν12 (503.6 cm−1), but the coupling remains unanalyzed. Anharmonic CCSD(T)/ANO1 estimates of the vibrational frequencies of syn formyl azide were in close agreement with previously published experimental and computational values. Experimentally determined vibration-rotation interaction (αi) values were in excellent agreement with coupled-cluster predicted αi values for the fundamentals ν9, ν12, and ν11.

































![1H-Thieno[2,3-c]pyrazole](http://img.cochemist.com/ccimg/25400/25337-79-5.png)
![1H-Thieno[2,3-c]pyrazole](http://img.cochemist.com/ccimg/25400/25337-79-5_b.png)

















![tricyclo[1.1.0.0~2,4~]butane](http://img.cochemist.com/ccimg/200/157-39-1.png)
![tricyclo[1.1.0.0~2,4~]butane](http://img.cochemist.com/ccimg/200/157-39-1_b.png)









![1-[3-(1-naphthyl)-5-(2-naphthyl)phenyl]naphthalene](http://img.cochemist.com/ccimg/173700/173678-08-5.png)
![1-[3-(1-naphthyl)-5-(2-naphthyl)phenyl]naphthalene](http://img.cochemist.com/ccimg/173700/173678-08-5_b.png)