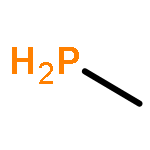
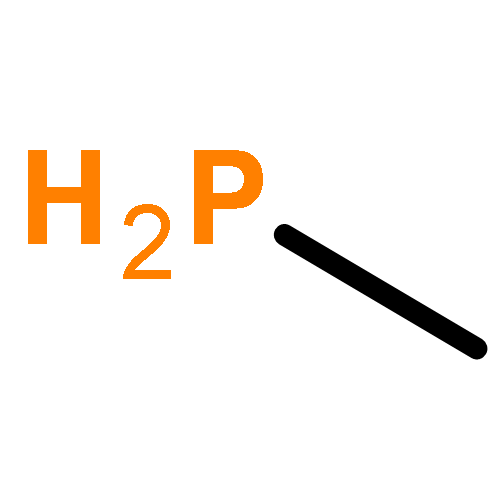
The Journal of Physical Chemistry A February 9, 2017 Volume 121(Issue 5) pp:1133-1139
Publication Date(Web):January 10, 2017
DOI:10.1021/acs.jpca.6b11610
To illustrate the formation mechanism of imidazolium-based ionic liquids (ILs) from N-alkyl imidazoles and halogenated hydrocarbons, density functional theory calculations have been carried out on a representative system, the reaction of N-methyl imidazole with chloroethane to form 1-ethyl-3-methyl imidazolium chloride ([Emim]Cl) IL. The reaction is shown to proceed via an SN2 transition state with a free energy barrier of 34.4 kcal/mol in the gas phase and 27.6 kcal/mol in toluene solvent. The reaction can be remarkably promoted by the presence of ionic products and water molecules. The calculated barriers in toluene are 22.0, 21.7, and 19.9 kcal/mol in the presence of 1–3 ionic pairs of [Emim]Cl and 23.5, 21.3, and 19.4 kcal/mol in the presence of 1–3 water molecules, respectively. These ionic pairs and water molecules do not participate directly in the reaction but provide a polar environment that favors stabilizing the transition state with large charge separation. Hence, we propose that the synthesis of imidazolium-based ILs from N-alkyl imidazoles and halogenated hydrocarbons is an autopromoted process and a polar microenvironment induced reaction, and the existence of water molecules (a highly polar solvent) in the reaction may be mainly responsible for the initiation of reaction.
The Journal of Physical Chemistry C April 16, 2009 Volume 113(Issue 15) pp:6215-6220
Publication Date(Web):2017-2-22
DOI:10.1021/jp8108489
While nanoscale gold particles show exceptional catalytic activity toward the water−gas shift (WGS) reaction, not much is known about the detailed reaction mechanism and the influence of charge state of Au nanoparticles on the reactivity. We here report a systematic theoretical study by carrying out density functional theory calculations for the WGS reaction promoted by cationic, neutral, and anionic Au dimers, which represent three simplest prototypes of Au nanoparticles with different charge states. The reaction mechanism is explored along two possible entrances: one involves the complexes of the dimers with CO and the other is related to the complexes of the dimers with H2O. In all cases, it is found that the catalytic cycle proceeds via the formate mechanism and involves two sequential elementary steps: the rupture of the O−H bond in H2O and the formation of H2 molecule. The calculated results show that the reaction mediated by Au2+ is energetically most favorable compared to those promoted by Au2 and Au2−, indicating that the charge state of Au dimers plays an essential role for the catalyzed WGS. The present theoretical study rationalizes the early experimental findings well and enriches our understanding of the catalytic WGS by Au-based catalysts.
Density functional theory calculations have been performed to understand the intriguing experimental observations on the hydration of propargylic alcohols to α-hydroxy ketones catalyzed by task-specific ionic liquids (ILs) and CO2. Focusing on a representative propargylic alcohol, 2-methylbut-3-yn-2-ol, we explored its hydration mechanism and the catalytic reactivities of different ILs towards the reaction in detail. The calculated results show the electrostatically controlled character of the reaction, where the reactivity depends on not only the anion's own nature but also its counterion cation that can regulate and control the anion basicity via electrostatic and H-bonding interactions. The reaction is proposed to proceed via an energetically viable mechanism that features the initial addition of CO2 to the hydroxyl group of the propargylic alcohol with assistance of the IL anion as a proton acceptor. The different catalytic performances of several ILs are attributed to their different proton-accepting capabilities. The best catalytic performance of [Bu4P][Im] is ascribed to its most efficient proton-accepting properties. The theoretical results provide a foundation for exploiting the controlled synthesis of α-hydroxy ketones as well as cyclic carbonates and oxazolidinones from the hydration of propargylic alcohols or propargylic amines.
Co-reporter:Xueli Mu, Xiaodeng Yang, Dongju Zhang, Chengbu Liu
Carbohydrate Polymers 2016 Volume 146() pp:46-51
Publication Date(Web):1 August 2016
DOI:10.1016/j.carbpol.2016.03.032
•DFT calculations illustrate the mechanism of the graft reaction of chitosan with the epoxy compound.•The polar environment of [Amim]Cl ionic liquid favors the graft reaction.•The ionic liquid also serves a catalyst of the reaction to promote the ring-opening of the epoxy compound.The molecular mechanism of the graft reaction of 2,3-epoxypropyl-trimethyl quaternary ammonium chloride with chitosan monomer was investigated by performing density functional theory (DFT) calculations. The calculated results show that the −NH2 group of chitosan monomer is more reactive than its −OH and −CH2OH groups, and the graft reaction on the −NH2 group is calculated to be exothermic by 20.5 kcal/mol with a free energy barrier of 42.6 kcal/mol. The reaction cannot benefit from the presence of the intruded water molecule, but can be considerably assisted by 1-allyl-3-methylimidazolium chloride ([Amim]Cl) ionic liquid. The reaction catalyzed by the ion-pair is calculated to be exothermic by 36.5 kcal/mol and the barrier is reduced to 29.3 kcal/mol, which are further corrected to 28.0 and 29.1 kcal/mol by considering the solvent effect of [Amim]Cl ionic liquid. Calculated results verified the experimental finding that imidazolium-based ionic liquids can promote the reaction of chitosan with epoxy compounds.
Co-reporter:Cuihuan Geng, Likai Du, Fang Liu, Rongxiu Zhu and Chengbu Liu
RSC Advances 2015 vol. 5(Issue 42) pp:33385-33391
Publication Date(Web):01 Apr 2015
DOI:10.1039/C4RA15202F
The selective fluorination of aromatic compounds with Selectfluor has been studied theoretically. The structural and energetic features of π complexes of substituted benzenes with Selectfluor are investigated, and the fluorine bond (F⋯π) has been found to make an important contribution to the stabilization of the π complexes. Our calculations indicate that the SET mechanism, which involves one electron transfer from the aromatic substrate (D) to Selectfluor (A), is preferred over the SN2 mechanism. The analysis of the minimum energy path (MEP) suggests that the DABCO moiety of Selectfluor seems to take an active role in the fluorination of aromatic compounds with Selectfluor. In addition, a two-state model analysis, as well as the characteristics of avoiding crossing between the DA and D+A− states of benzene/Selectfluor are addressed to obtain deep insight into the features of the SET mechanism.
Co-reporter:Cuihuan Geng, Sujuan Zhang, Chonggang Duan, Tongxiang Lu, Rongxiu Zhu and Chengbu Liu
RSC Advances 2015 vol. 5(Issue 97) pp:80048-80056
Publication Date(Web):15 Sep 2015
DOI:10.1039/C5RA16359E
The mechanisms of Selectfluor-mediated Au-catalyzed intramolecular Csp3–Csp2 cross-coupling reaction involving direct aryl Csp2–H functionalization have been investigated theoretically. Several pathways involving the oxidation of alkylgold(I) (Cycle I), phosphine Au(I) precatalyst (Cycle II), gold(I) π–alkene complex (Cycle III) and arylgold(I) (Cycle IV) by Selectfluor, respectively, were examined. Our calculation results suggested the following: (1) Cycles I and II are preferred over Cycles III and IV, and the reaction would undergo the energy favored pathways (Cycles I and II), which is further confirmed by stereochemical analysis; (2) Cycle I is competitive with Cycle II, and the rate-determining steps of these two cycles are oxidation of Au(I) species by Selectfluor; (3) water has been found to participate in the catalytic reaction and decrease the activation energy barrier of the reductive elimination.
Co-reporter:Cuihuan Geng;Rongxiu Zhu;Mingxia Li;Tongxiang Lu; Steven E. Wheeler; Chengbu Liu
Chemistry - A European Journal 2014 Volume 20( Issue 48) pp:15833-15839
Publication Date(Web):
DOI:10.1002/chem.201404277
Abstract
The pairing of transition metal catalysis with the reagent Selectfluor (F-TEDA–BF4) has attracted considerable attention due to its utility in myriad CC and Cheteroatom bond-forming reactions. However, little mechanistic information is available for Selectfluor-mediated transition metal-catalyzed reactions and controversy surrounds the precise role of Selectfluor in these processes. We present herein a systematic investigation of homogeneous Au-catalyzed oxidative CO bond-forming reactions using density functional theory calculations. Currently, Selectfluor is thought to serve as an external oxidant in AuI/AuIII catalysis. However, our investigations suggest that these reactions follow a newly proposed mechanism in which Selectfluor functions as an electrophilic fluorinating reagent involved in a fluorination/defluorination cycle. We have also explored Selectfluor-mediated gold-catalyzed homocoupling reactions, which, when cyclopropyl propargylbenzoate is used as a substrate, lead to an unexpected byproduct.
Computational studies to determine the origin of enantioselectivity in the (1R,2R)-1,2-diphenylethane-1,2-diamine (DEPN)–Brønsted acid catalyzed epoxidation of 2-cyclohexen-1-one have been performed using density functional theory. Transition states for conjugate addition and ring closure steps of the epoxidations catalyzed by three different catalyst systems were characterized. Our calculations show that the Csp2H⋯O H-bond interaction between the benzene ring of the catalyst and H2O is mainly responsible for the chiral discrimination observed. The Brønsted acid counterion plays a very important role in ensuring high enantioselectivity by improving the rigidity of the transition state structures to allow the efficient formation of the Csp2H⋯O H-bond. Moreover, we explain why these two diamine catalysts (1S,2S)-DACH and (1R,2R)-DPEN display consistent enantioselectivities in the catalytic epoxidation of 2-cyclohexen-1-one when combining with three different cocatalysts; achiral TFA, and chiral (R)- and (S)-TRIP.
By employing ab initio quantum mechanical/molecular mechanical (QM/MM) and molecular dynamics (MD) simulations, we have provided further evidence against the previously proposed hydroperoxylation or hydroxylation mechanism of hydroxyethylphosphonate dioxygenase (HEPD). HEPD employs an interesting catalytic cycle based on concatenated bifurcations. The first bifurcation is based on the abstraction of hydrogen atoms from the substrate, which leads to a distal or proximal hydroperoxo species (Fe–OOH or Fe–(OH)O). The second and the third bifurcations refer to the carbon–carbon bond cleavage reaction. And this is achieved through a tridentate intermediate, or employing a proton-shuttle assisted mechanism, in which the residue Glu176 or the FeIVO group serves as a general base. The reaction directions seem to be tunable and show significant environment dependence. This mechanism can provide a comprehensive interpretation for the seemingly contradicting experimental evidences and provide insight into the development of biochemistry and material sciences.
Co-reporter:Xueying Zhu, Jianqiang Liu, Dongju Zhang, Chengbu Liu
Computational and Theoretical Chemistry 2012 Volume 996() pp:21-27
Publication Date(Web):15 September 2012
DOI:10.1016/j.comptc.2012.07.008
By performing density functional theory calculations, we systematically studied the Diels–Alder (D–A) reaction between cyclopentadiene and methacrylate catalyzed by alanine methyl ester nitrate ([AME][NO3]), an amino acid-based ionic liquid (AAIL). The uncatalyzed reaction was first calculated in both gas phase and dichloromethane, and then the catalytic effect of [AME][NO3] AAIL on the D–A reaction was mimicked by using one, two, and up to three ion pairs as catalysts. The calculated results show that [AME][NO3] plays a role of Lewis acid to promote the reaction and the catalytic active center is the NH3 group in [AME]+ cation, which forms the NH⋯O H-bond with the carbonyl oxygen atom in methacrylate to effectively polarize the CC double bond in methacrylate. As a result, the energy barrier of reaction is remarkably reduced, and the asynchronicity of reaction is increased. The calculated energy barrier for the reaction with the presence of two ion pairs is lower than those with the presences of one and three ion pairs, implying that the optimal molar ratio among two reactants and the reaction medium/catalyst [AME][NO3] should be 1:1:2. The present results rationalize the early experimental findings, and provide a useful reference for the rational design of usual D–A reactions in AAILs.Graphical abstractHighlights► The [AME][NO3]-catalyzed Diels–Alder reaction between cyclopentadiene and methacrylate is studied theoretically. ► [AME][NO3] plays a role of Lewis acid to promote the reaction. ► The catalytic active center is the NH3 group in [AME]+ cation. ► The optimal molar ratio among two reactants and the catalyst may be 1:1:2.
Co-reporter:Likai Du, Jun Gao, Yongjun Liu, and Chengbu Liu
The Journal of Physical Chemistry B 2012 Volume 116(Issue 39) pp:11837-11844
Publication Date(Web):September 5, 2012
DOI:10.1021/jp305454m
The hydroxyethylphosphonate dioxygenase (HEPD) catalyzes the critical carbon–carbon bond cleavage step in the phosphinothricin (PT) biosynthetic pathway. The experimental research suggests that water molecules play an important role in the catalytic reaction process of HEPD. This work proposes a water involved reaction mechanism where water molecules serve as an oxygen source in the generation of mononuclear nonheme iron oxo complexes. These molecules can take part in the catalytic cycle before the carbon–carbon bond cleavage process. The properties of trapped water molecules are also discussed. Meanwhile, water molecules seem to be responsible for converting the reactive hydroxyl radical group (−OH) to the ferric hydroxide (Fe(III)–OH) in a specific way. This converting reaction may prevent the enzyme from damages caused by the hydroxyl radical groups. So, water molecules may serve as biological catalysts just like the work in the heme enzyme P450 StaP. This work could provide a better interpretation on how the intermediates interact with water molecules and a further understanding on the O18 label experimental evidence in which only a relatively smaller ratio of oxygen atoms in water molecules (∼40%) are incorporated into the final product HMP.
Co-reporter:Qiang Wang, Jun Gao, Yongjun Liu, Chengbu Liu
Journal of Molecular Graphics and Modelling 2012 Volume 38() pp:186-193
Publication Date(Web):September 2012
DOI:10.1016/j.jmgm.2012.06.011
The protein tyrosine phosphatase 1B (PTP-1B) is acknowledged as an outstanding therapeutic target for the treatment of diabetes, obesity and cancer. In this work, six aryl diketoacid compounds have been studied on the basis of molecular dynamics simulations. Hydrogen bonds, binding energies and conformation changes of the WPD loop have been analyzed. The results indicated that their activation model falls into two parts: the target region of the monomeric aryl diketoacid compounds is the active site, whereas the target region of the dimeric aryl diketoacid compounds is the WPD loop or the R loop. The van der Waals interactions exhibit stronger effects than the short-range electrostatic interactions. The van der Waals interaction energy and the IC50 values exhibit an approximately exponential relationship. Furthermore, the van der Waals interactions cooperate with the hydrogen bond interactions. This study provides a more thorough understanding of the PTP-1B inhibitor binding processes.Graphical abstractHighlights► Monomeric and dimeric aryl diketoacids adopt different bonding modes. ► The van der Waals interaction plays an important role. ► The van der Waals interaction energy and their IC50 values are closely correlated. ► The van der Waals and the hydrogen bond interaction are mutually cooperative.
Co-reporter:Ping-Li Lv, Rong-Xiu Zhu, Dong-Ju Zhang, Chong-Gang Duan, and Cheng-Bu Liu
The Journal of Physical Chemistry A 2012 Volume 116(Issue 4) pp:1251-1260
Publication Date(Web):December 27, 2011
DOI:10.1021/jp207914h
The asymmetric epoxidation of 2-cyclohexen-1-one with aqueous H2O2 as oxidant, 1,2-diaminocyclohexane as catalyst, and a Brønsted acid trifluoroacetic acid (TFA) as cocatalyst has been studied by performing density functional theory calculations. It is confirmed that the catalyzed epoxidation proceeds via sequential nucleophilic addition and ring-closure processes involving a ketiminium intermediate. Four possible pathways associated with two Z isomers and two E isomers of ketiminium have been explored in detail. Our calculation indicates that these four pathways have high barriers and a small energy gap between two more favorable R and S pathways. We have analyzed the effects of the TFA anion and H2O on the activity and enantioselectivity of catalytic epoxidation. It is found that the TFA anion acts as a counterion to stabilize the transition states of the catalytic epoxidation by hydrogen–bond acceptance, leading to decreases in the barriers of the nucleophilic addition and ring-closure processes. The most significant decrease occurred in the ring-closure step of the Z-R-pathway, resulting in H-bond-induced enantioselectivity. Our calculations also show that water cooperates with TFA to further increase the reaction rate significantly.
Co-reporter:Xin Che, Jun Gao, Dongju Zhang, and Chengbu Liu
The Journal of Physical Chemistry A 2012 Volume 116(Issue 22) pp:5510-5517
Publication Date(Web):May 15, 2012
DOI:10.1021/jp3001515
In the iron(II)-thiolate models of cysteine dioxygenase, the thiolate ligand is a key factor in the oxygen activation. In this contribution, four model compounds have been theoretically investigated. This comparative study reveals that the thiolate ligand itself and its relative position are both important for the activation of O2. Before the O2 binding, the thiolate ligand must transfer charge to Fe(II), and the effective nuclear charges of Fe(II) is decreased, which results in a lower redox potential of compounds. In other words, the thiolate ligand provides a prerequisite for the O2 activation. Furthermore, the relative position of the thiolate ligand is discovered to determine the reaction path of O2 activation. The amount of charge transfer is crucial for these reactions; the more charge it transfers, the lower the related redox potentials. This work really helps think deeper into the O2 activation process of mononuclear nonheme iron enzymes.
Co-reporter:Zhen Xu, Ke Song, Shi-Ling Yuan, and Cheng-Bu Liu
Langmuir 2011 Volume 27(Issue 14) pp:8611-8620
Publication Date(Web):June 3, 2011
DOI:10.1021/la201328y
Molecular dynamics simulations are used to study the micronature of the organization of water molecules on the flat surface of well-ordered self-assembled monolayers (SAMs) of 18-carbon alkanethiolate chains bound to a silicon (111) substrate. Six different headgroups (−CH3, −C═C, −OCH3, −CN, −NH2, −COOH) are used to tune the character of the surface from hydrophobic to hydrophilic, while the level of hydration is consistent on all six SAM surfaces. Quantum mechanics calculations are employed to optimize each alkyl chain of the different SAMs with one water molecule and to investigate changes in the configuration of each headgroup under hydration. We report the changes of the structure of the six SAMs with different surfaces in the presence of water, and the area of the wetted surface of each SAM, depending on the terminal group. Our results suggest that a corrugated and hydrophobic surface will be formed if the headgroups of SAM surface are not able to form H-bonds either with water molecules or between adjacent groups. In contrast, the formation of hydrogen bonds not only among polar heads but also between polar heads and water may enhance the SAM surface hydrophilicity and corrugation. We explicitly discuss the micromechanisms for the hydration of three hydrophilic SAM (CN-, NH2- and COOH-terminated) surfaces, which is helpful to superhydrophilic surface design of SAM in biomimetic materials.
Co-reporter:Xueying Zhu, Peng Cui, Dongju Zhang, and Chengbu Liu
The Journal of Physical Chemistry A 2011 Volume 115(Issue 29) pp:8255-8263
Publication Date(Web):June 14, 2011
DOI:10.1021/jp201246j
By performing density functional theory calculations, we have studied the synthesis mechanism, electronic structure, and catalytic reactivity of a pyridinium-based ionic liquid, 1-ethylpyridinium trifluoroacetate ([epy]+[CF3COO]−). It is found that the synthesis of the pyridinium salt follows a SN2 mechanism. The electronic structural analyses show that multiple H bonds are generally involved in the pyridinium-based ionic liquid, which may play a decisive role for stabilizing the ionic liquid. The cation–anion interaction mainly involves electron transfer between the lone pair of the oxygen atom in the anion and the antibonding orbital of the C*–H bond (C* denotes the carbon atom at the ortho-position of nitrogen atom in the cation). This present work has also given clearly the catalytic mechanism of [epy]+[CF3COO]− toward to the Diels–Alder (D-A) reaction of acrylonitrile with 2-methyl-1,3-butadiene. Both the cation and anion are shown to play important roles in promoting the D-A reaction. The cation [epy]+, as a Lewis acid, associates the C≡N group by C≡N···H H bond to increase the polarity of the C═C double bond in acrylonitrile, while the anion CF3COO– links with the methyl group in 2-methyl-1,3-butadiene by C–H···O H bond, which weakens the electron-donating capability of methyl and thereby lowers the energy barrier of the D-A reaction. The present results are expected to provide valuable information for the design and application of pyridinium-based ionic liquids.
Journal of Molecular Modeling 2011 Volume 17( Issue 8) pp:1997-2004
Publication Date(Web):2011 August
DOI:10.1007/s00894-010-0879-1
To better understand the property of the binary systems composing of imidazolium salt, [emim]+Aˉ (A=Clˉ, Brˉ, BF4ˉ, and PF6ˉ) and methanol, we have investigated in detail the interactions of methanol molecule with anions Aˉ, cation [emim]+, and ion pair [emim]+Aˉ of several ionic liquids (ILs) based on 1-ethyl-3-methylimidazolium cation by performing density functional theory calculations. It is found that H-bonds are universally involved in these systems, which may play an important role for the miscibility of methanol with imidazolium-based ILs. The interaction mechanisms of methanol molecule with anion and cation are found to be different in nature: the former mainly involves LPX-\( \sigma_{{O - H}}^{*} \) interaction, while the latter relates with the decisive orbital overlap of the type of LPO-\( \sigma_{{C - H}}^{*} \). Based on the present calculations, we have provided some reasonable interpretations for properties of the binary mixtures of ILs and alcohol and revealed valuable information for the interaction details between ILs and alcohols, which is expected to be useful for the design of more efficient ILs to form superior solvent system with alcohol.
Magnetic coupling interactions of a MnIII4 system are investigated by calculations based on density functional theory combined with a broken-symmetry approach (DFT-BS). Three different interactions including ferromagnetic and antiferromagnetic coupling are concomitant in this complex. This magnetic phenomenon of the complex is due to the different bridging angles between the Mn(III) centers in the three different models and the orbital complementarity of the μ-pzbg and μ-OCH3 bridging ligands, which is proven by the analyses of the molecular orbitals. According to the analyses of the magneto-structural correlation, it is revealed that the magnetic coupling interaction switches from ferromagnetic to antiferromagnetic at the point of the bridging angle Mn–(μ-OCH3)–Mn = 99°, which is equal to the value in the origin crystal. Significant correlation between the magnetic properties and the component of the d orbitals in these systems shows that the larger contribution of the dz2 orbital corresponds to the larger ferromagnetic coupling interaction. These results should provide a means to control the magnetic coupling of the polynuclear Mn systems, which is instructive for the design of new molecular magnetic materials.
Co-reporter:Jinxin Guo, Dongju Zhang, Chonggang Duan, Chengbu Liu
Carbohydrate Research 2010 Volume 345(Issue 15) pp:2201-2205
Publication Date(Web):13 October 2010
DOI:10.1016/j.carres.2010.07.036
The interactions of the cellulose molecule with several anions, including acetate , alkyl phosphate, tetrafluoroborate and hexafluorophosphate anions which are most commonly involved in the imidazolium ionic liquids (ILs), have been studied by performing density functional theory calculations. Based on calculated geometries, energies, IR characteristics, and electronic properties of the cellulose–anion complexes, it is found that the strength of interactions of anions with cellulose follows the order: acetate anion > alkyl phosphate anion > tetrafluoroborate anion > hexafluorophosphate anion, which is consistent with the experimentally observed solubility trend of cellulose in the corresponding imidazolium-based ILs. The present study may provide basic aids to some extent for understanding the dissolution behavior of cellulose in the imidazolium-based ILs.
Co-reporter:Zhang-Yu Yu, Tao Liu, Dao-Jun Guo, Yong-Jun Liu, Cheng-Bu Liu
Journal of Molecular Structure 2010 Volume 984(1–3) pp:402-408
Publication Date(Web):15 December 2010
DOI:10.1016/j.molstruc.2010.10.015
The microenvironmental effect of dimethyl sulfoxide (DMSO) on adrenaline was studied by several approaches including the cyclic voltammetry (CV) of adrenaline at a platinum electrode in acid aqueous solution, the chemical shift of 1H nuclear magnetic resonance (1H NMR) of adrenaline, and the change of diffusion coefficient of adrenaline. The experimental results demonstrated that DMSO has significant microenvironmental effect on adrenaline, which was confirmed by the density functional theory (DFT) study on the hydrogen bond (H-bond) complexes of adrenaline with water and DMSO.
Co-reporter:Qiang Wang, Jun Gao, Yongjun Liu, Chengbu Liu
Chemical Physics Letters 2010 Volume 501(1–3) pp:113-117
Publication Date(Web):6 December 2010
DOI:10.1016/j.cplett.2010.10.048
Abstract
A ribosome is a large RNA–protein machine that catalyzes protein synthesis. RNA plays a crucial role in catalyzing peptide bond formation. In this Letter, we focus on the catalytic role of the A76 2′-OH of the P site in tRNA. Three possible reaction schemes were compared using the same reaction model and theoretical level. By comparison of the potential energy surface and optimized geometries of transition states, a new favorable reaction pathway is shown. The activation energy is 24.0 kcal/mol. This work will provide a foundation for further theoretical and experimental study of ribosome mechanisms.
Journal of Molecular Structure: THEOCHEM 2010 Volume 948(1–3) pp:78-82
Publication Date(Web):30 May 2010
DOI:10.1016/j.theochem.2010.02.025
The polypeptide-mediated electron transfers (ET) in opposite directions are studied by analyzing the electronic structures. The electron delocalizations in polypeptides are mainly occurring from carboxyl end to amino end (direction A). The lowest unoccupied molecular orbital (LUMO) energies of the peptide subgroups decrease obviously along this direction. Accordingly, the ET in direction A would occur by the superexchange mechanism. The formation of intramolecular hydrogen bond may provide new tunneling pathway with lower energy barrier, and promotes the ET in direction A markedly. As to the ET in opposite direction, the thermally activated hopping mechanism should be adopted.
Co-reporter:Zhangyu Yu, Tao Liu, Dongju Zhang, Chengbu Liu
Journal of Molecular Structure: THEOCHEM 2010 Volume 960(1–3) pp:10-14
Publication Date(Web):30 November 2010
DOI:10.1016/j.theochem.2010.08.017
The hydrogen bond (H-bond) interaction of 1:1 supermolecular complexes of protonated adrenaline (PAd+) with formate anion and its derivatives (denoted as RCOO−, RH, CH3, CH2F, CH2Cl, and CH2Br) has been investigated by performing density functional theory calculations at the B3LYP/6-31G+(d) level. We obtained the most stable three conformations for each complex, which are denoted as PAd+–RCOO−(I), PAd+–RCOO−(II), PAd+–RCOO−(III), respectively, and calculated the interaction energy between PAd+ and RCOO−. In all PAd+–RCOO− complexes, PAd+–CH3COO− is found to be the most favorable energetically. There exists low-barrier hydrogen bond (LBHB) in PAd+–HCOO−(III), PAd+–CH2FCOO−(III), PAd+–CH2ClCOO−(III), and PAd+–CH2Br−(III) complexes. The solvent effects on the geometry and energy of the complexes are also considered by using the polarizable continuum model (PCM) model in aqueous solvent. It is found that PAd+–R− complexes in solution are significantly less stable than those in the gas-phase. The theoretical results for the present model systems will be useful for experimental researchers working in this field.
Co-reporter:Rong-Xiu Zhu, Dong-Ju Zhang, Jin-Xin Guo, Jing-Lin Mu, Chong-Gang Duan and Cheng-Bu Liu
The Journal of Physical Chemistry A 2010 Volume 114(Issue 13) pp:4689-4696
Publication Date(Web):March 15, 2010
DOI:10.1021/jp100291c
A computational study with the B3LYP density functional theory was carried out to study the reaction mechanism for the cycloisomerization of allenes catalyzed by Au(I) and Au(III) complexes. The catalytic performance of Au complexes in different oxidation states as well as the effects of the counterion on the catalytic activities has been studied in detail. Our calculations show that the catalytic reaction is initiated by coordination of the Au(I) or Au(III) catalyst to the distal double bond of allene and activation of allene toward facile nucleophilic attack, then 3-pyrroline obtained via two-step proton shift, followed by demetalation. On the basis of our calculations, H shifts are key steps of the catalytic cycle, which are significantly affected by the gold oxidation state, counterion, ligands, and assistant catalyst. AuCl is found to be more reactive than AuCl3; however, the Au(III)-catalyzed path does not involve an oxidation state change from Au(III) to Au(I). Our calculated results rationalize the experimental findings well and overthrow the previous conjecture about Au(I) serving as the catalytically active species for Au(III)-catalyzed cycloisomerization.
Co-reporter:Yingying Wang, Dongju Zhang, Zhangyu Yu and Chengbu Liu
The Journal of Physical Chemistry C 2010 Volume 114(Issue 6) pp:2711-2716
Publication Date(Web):January 26, 2010
DOI:10.1021/jp9103596
Density functional theory calculations have been performed to elucidate the mechanism of N2O formation over the Au(111) surface during NO reduction. It is shown that the dissociation of NO into an N atom and an O atom involves a barrier as high as 3.9 eV, implying that the formation of N2O does not occur via the direct dissociation mechanism of NO. Alternatively, we find that the reaction may occur via a dimer mechanism; i.e., two NO molecules initially associate into a dimeric (NO)2, which then dissociates into a N2O molecule and a N atom. We have scanned the potential energy surface forming N2O along different pathways, which involve a trapezoid OadNNOad dimer, an inverted trapezoid ONadNadO dimer, a zigzag ONadNOad dimer, or a rhombus ONadOadN dimer. The trapezoid dimer, OadNNOad, is found to be a necessary intermediate for the formation of N2O, and the calculated barrier for the rate-determining step along the energetically most favorable pathway is only 0.34 eV. The present results rationalize the early experimental findings well and enrich our understanding of the reduction of NO on the Au surface.
Co-reporter:Guiqing Zhang, Peng Cui, Jian Wu, Chengbu Liu
Chemical Physics Letters 2009 Volume 471(1–3) pp:163-167
Publication Date(Web):16 March 2009
DOI:10.1016/j.cplett.2009.02.009
Abstract
Segments of the DNA are calculated with the Peyrard–Bishop–Holstein model. The results show that the two controversial charge transport mechanisms, hopping and tunneling, are not consummate to depict its behaviors along the DNA chains composed of , in which the polaron will annihilate gradually and meanwhile new one will be created at some new ones in the presence of an electric field. We speculate the phenomenon can be explained by the collective vibration of the lattices and the two intensity couplings: one is between lattices and a charge; the other is hydrogen bond interaction between complementary bases.
Co-reporter:Zhe Han, Dongju Zhang, Youmin Sun, Chengbu Liu
Chemical Physics Letters 2009 Volume 474(1–3) pp:62-66
Publication Date(Web):25 May 2009
DOI:10.1016/j.cplett.2009.04.044
Abstract
By performing DFT calculations, the reaction of 4-chlorophenol with OH is reexamined to reconcile an experimental finding with a recent theoretical result. The calculated results show that abstracting hydrogen atom in the hydroxyl of 4-chlorophenol by the OH is the most plausible process for forming 4-chlorocaechol intermediate, while adding OH to the aromatic ring is the dominant pathway for forming the hydroquinone. The 4-chlorophenoxyl radical plays a crucial role during the hydroxyl-initiated 4-CP degradation due to its energetic stability and the low barrier involved in the reaction, which supports the experimental finding but differs from the recent theoretical result.
The Journal of Physical Chemistry B 2009 Volume 113(Issue 30) pp:10399-10402
Publication Date(Web):July 2, 2009
DOI:10.1021/jp903835j
The glycine zwitterion (GlyA) in the gas phase is not a local energy minimum and transforms to the canonical isomer (GlyB) via a barrierless process. Within ZSM-5 zeolite, it is rendered geometrically stable and even has a lower energy of 7.57 kcal mol−1 than GlyC, the most stable isomer of glycine in the gas phase; GlyB represents the lowest energy minimum, which is facile to transform into the zwitterion with low-energy barrier (4.46 kcal mol−1). In addition, the zwitterion can be efficiently obtained by adsorption of glycine in the deprotonated form at the acidic site of HZSM-5 zeolite. The relative stability of glycine isomers in silicalite-1 increases in the order GlyA < GlyB < GlyC, the same as that in the gas phase. Silicalite-1 stabilizes GlyA somewhat, whereas it destabilizes GlyB greatly. The negative charges of ZSM-5 zeolite created by Al doping are indispensable to the stabilizations of the zwitterion; however, the lattices also play an important role and approximate 74.1% of the contributions of the negative charges.
Co-reporter:Xiaofeng Wei, Dongju Zhang, Chengbu Liu
Journal of Molecular Structure: THEOCHEM 2009 Volume 909(1–3) pp:1-5
Publication Date(Web):15 September 2009
DOI:10.1016/j.theochem.2009.05.016
We have carried out a detailed theoretical study for the geometrical structures and electronic properties of the cation and the ion pair of the cycle tetramethylguanidinium nitrate ionic liquid. It is found that in the cation, the triazolium ring presents almost a plane and the NMe2 group is not coplanar with the ring. The central C2 atom carries high positive charge, which is expected to act as the most active site for the electrostatic attacking of an anion. For the ion pair, three most stable configurations have been located, where the anion lies on either side of the triazolium ring or between C5 and C6 in the cation via multiple hydrogen bonds. However, the calculated results show that the electrostatic interaction between cation and anion plays a crucial role for stabilizing the ion pair. Moreover, the charge-localized character of the cation effectively increases the electrostatic interaction between the cation and anion, which is mainly responsible for the high thermal stabilities of the ionic liquids. There is a close correlation between the charge on C2 and the relative stability of the ion pair: the more positive charge on C2, the more stable the ion pair. Natural bond orbital and frontier molecular orbital analyses reveal that the charge transfer from the anion to cation occurs mainly through the LPO→σC–H∗, LPO→πC2–N3∗ or LPO→πC2–N1∗ interactions.
Co-reporter:Ye-Fei Wang, Zhang-Yu Yu, Jian Wu and Cheng-Bu Liu
The Journal of Physical Chemistry A 2009 Volume 113(Issue 39) pp:10521-10526
Publication Date(Web):September 4, 2009
DOI:10.1021/jp9020036
In this work, the electron structure and charge-transfer mechanism in polypeptide chains are investigated according to natural bond orbitals (NBO) analysis at the level of B3LYP/6-311++G**. The results indicate that the delocalization of electrons between neighboring peptide subgroups can occur in two opposite directions, and the delocalization effect in the direction from the carboxyl end to the amino end has an obvious advantage. As a result of a strong hyperconjugative interaction, the lowest unoccupied NBO of the peptide subgroup, π*C−O, has significant delocalization to neighboring subgroups, and the energies of these NBOs decrease from the carboxyl end to the amino end. The formation of intramolecular O···H−N type hydrogen bonds also helps to delocalize the electron from the carboxyl end to the amino end. Thus, the electron will flow to the amino end. The superexchange mechanism is suggested in the electron-transfer process.
Co-reporter:Li-Li Wang, You-Min Sun, Zhang-Yu Yu, Zhong-Nan Qi and Cheng-Bu Liu
The Journal of Physical Chemistry A 2009 Volume 113(Issue 39) pp:10534-10539
Publication Date(Web):September 8, 2009
DOI:10.1021/jp9045897
The mechanisms of the magnetic coupling interactions for two trigonal−bipyramid trinuclear Cu(II) complexes Cu3(μ3-X)2(μ-pz)3X3 (X = Cl and Br, respectively) and three trigonal trinuclear Cu(II) complexes Cu3(μ3-X)(μ-pz)3Cl3 (X = Cl, Br, and O) are investigated by the calculations based on density functional theory combined with broken-symmetry approach (DFT-BS). The research on the magneto-structural correlation reveals that the magnetic coupling interaction is sensitive to the Cu-(μ3-X)-Cu angle. With the Cu-(μ3-X)-Cu angle changing from 76 to 120°, the magnetic coupling interaction is switched from ferromagnetic to antiferromagnetic. According to the analysis of the molecular orbitals and the variation of the spin-state energies versus the ratio of the magnetic coupling constants, it is found that there exists spin frustration phenomenon in these complexes.
Journal of Molecular Structure: THEOCHEM 2009 Volume 901(1–3) pp:81-87
Publication Date(Web):15 May 2009
DOI:10.1016/j.theochem.2009.01.006
B3LYP and MP2 theoretical methods were employed to study the structures and relative stabilities of the Gly, Ala, Val, Aib, F-Gly, I-Gly and dF-Gly conformers. It was found that the relative stabilities of the Gly, Ala, Val and Aib conformers increase in the same orders of V < IV < III < II < I, due to the similar shapes of the corresponding conformers. Two exceptions exist for the above orders: In Gly, conformer IV is slightly more stable than conformer III; in Aib, conformer II is more stable than conformer I. The relative stability of conformer II vs. I is gradually enhanced by the substitutions of the lateral H atoms with the alkyl groups, especially in the case of the double substitutions to form Aib. The geometries and relative stabilities of the F-Gly, I-Gly and dF-Gly conformers are quite different from Gly as well as Ala, Aib and Val with the alkyl side chains. The driving forces in the conformers of the halogenated amino acids are mainly the intramolecular hydrogen bonding interactions, at least four hydrogen bonds formed in each dF-Gly conformer. For F-Gly and I-Gly, three conformers II–IV exist whereas conformers I and V are spontaneously transformed into conformers IV and III, respectively. It was found that the functional rather than alkyl side chains interact strongly with the NH2CHCOOH fragments and accordingly have more remarkable influences on the geometries and relative stabilities of the conformers.
Co-reporter:Xiaofeng Wei, Dongju Zhang, Chengbu Liu
Journal of Molecular Structure: THEOCHEM 2008 Volume 859(1–3) pp:1-6
Publication Date(Web):30 June 2008
DOI:10.1016/j.theochem.2008.02.020
The reactivity of silica toward to water is an important issue in environmental science and materials science for many years. In this paper, we study the hydrolysis stabilities of silica molecular chains and molecular rings based on two-membered silica ring by performing density functional theory calculations. It is found that the hydrolysis for the linear molecular chains firstly takes place in the middle parts and carries on gradually to the ends. In contrast with the early conjecture that the molecular rings might have higher hydrolysis stability than the corresponding linear chains, we find that these fully coordinated molecular rings are less stable than the corresponding molecular chains with the presence of water molecule.
Science China Chemistry 2008 Volume 51( Issue 12) pp:1182-1186
Publication Date(Web):2008 December
DOI:10.1007/s11426-008-0128-y
The polaron might play an important role in the process of charge migration through duplex DNA stack. In the present work, we investigate properties of hole polarons in DNA molecules containing identical base pairs, such as poly(G)-poly(C) and poly(A)-poly(T), An extended tight-binding model (extended Su-Schrieffer-Heeger model), which involves the effect of an electric field in the direction of DNA stack, will be introduced. The transfer integral and electron-phonon coupling parameters in this model are obtained according to ab initio calculation for different base pair dimers. Calculations reveal that the polaron in poly(A)-poly(T) has a wider shape and a higher mobility under a specific electric field than that in poly(G)-poly(C) DNA stack.
Co-reporter:Li-Li Wang, You-Min Sun, Zhong-Nan Qi and Cheng-Bu Liu
The Journal of Physical Chemistry A 2008 Volume 112(Issue 36) pp:8418-8422
Publication Date(Web):August 19, 2008
DOI:10.1021/jp8044252
The mechanisms of magnetic exchange interactions in two heterobridged μ-hydroxyl-μ-X dicopper complexes A and B (X = azaindole for A and X = pyrazole for B) are investigated by the calculations based on density functional theory combined with the broken-symmetry approach (DFT-BS). It is found that although the coordination circumstances of the copper centers in the two complexes are very similar, the magnetic magnitudes and signs are diametrically opposed. By the theoretical analyses of magnetic orbital interaction and spin distribution, it is indicated that the difference between the magnetic properties of the two complexes is due to the distinction of orbital interaction of two bridge ligands. Namely, the weak ferromagnetic coupling for complex A arises from the orbital countercomplementarity of the hydroxo and azaindole bridges while the strong antiferromagnetic coupling for complex B arises from the orbital complementarity of the hydroxo and pyrazolato bridges.
The Journal of Physical Chemistry C 2008 Volume 112(Issue 43) pp:16729-16732
Publication Date(Web):October 4, 2008
DOI:10.1021/jp807264n
We show by performing density functional theory calculations that the dimer of titanium dioxide (TiO2) molecule, Ti2O4, is qualified for serving as a basic building block of TiO2 nanostructures owing to its structural stability and appropriate growth activity. In particular, the two thinnest titanium dioxide nanowires, as the prototypes of TiO2 nanostructures, have been assembled and proved to be geometrically graceful and dynamically stable. Calculated results show that the size and shape of TiO2 nanowires have important effects on their structural stabilities and energy gaps, proposing that tailoring the size and shape of TiO2 nanowires may be an effective way to modulate the band gap and finally improve their optical properties.
Co-reporter:Ruoxi Wang, Dongju Zhang, Rongxiu Zhu, Chengbu Liu
Journal of Molecular Structure: THEOCHEM 2007 Volume 817(1–3) pp:119-123
Publication Date(Web):1 September 2007
DOI:10.1016/j.theochem.2007.04.025
The geometrical structures and relative stabilities of boron-rich boron carbon clusters BnC (n = 1–7) are investigated using density functional theory calculations. The planar multi-cyclic geometries for n = 3–7 with C atom at the apex with most B–C bonds and most three-coordination boron atoms are the most stable structures. It is interesting that the C atoms reside in the apexes not doped in the B rings, that is the doped C atoms prefer to the surface of the B structures and do not change the frame structures of B clusters. The compact 3D structures and the linear isomers, however, are energetically more unfavorable for n = 3–7 with the increase of the size. The calculated disproportionation energy, binding energy and HOMO–LUMO gaps show that the B2C and B6C clusters are magic clusters.
Co-reporter:Rongxiu Zhu, Dongju Zhang, Jian Wu, Chengbu Liu
Journal of Molecular Structure: THEOCHEM 2007 Volume 815(1–3) pp:105-109
Publication Date(Web):1 August 2007
DOI:10.1016/j.theochem.2007.03.025
The tandem nitroso aldol/Michael reaction between nitrosobenzene and cyclohexenone with pyrrolidine-based catalyst has been recently reported to obtain O-nitroso Diels–Alder bicyclic products. We present here a theoretical study for the novel reaction to rationalize the experimental findings of the regioselectivity and bicyclic products of the reaction. By performing density functional theory calculations, we have identified the detailed mechanism of the title reaction and the pivotal factors controlling the regioselectivity of the reaction. Two regioselective channels (O- and N-selective) for the aldol/Michael reaction have been characterized in detail. The calculated results indicate that both the aldol reaction and the next Michael reaction for the O-selective channel are much more favorable in energy than the corresponding N-selective channel. Theoretical results account well for the regioselectivity and the formal nitroso Diels–Alder adducts observed in the recent experiment.
The direct catalytic enantioselective α-amination reaction of carbonyl compounds, a powerful approach to asymmetric carbon–nitrogen bond-forming, has been extensively studied, however, our understanding of the mechanism is far from complete. A theoretical study is presented for the α-amination reaction of 2-acetylcyclopentanone with azodicarboxylate catalyzed by a urea-based chiral bifunctional organocatalyst. By performing density functional theory (DFT) calculations, we have identified a detailed mechanism of the reaction and the roles of the amino group and urea in the catalyst. The structures of the catalyst, substrates, intermediates, and transition states involved in the reaction have been located along four possible reaction channels. The rate-determining step is the C–N bond-forming step. The calculations show that the catalyst promotes the reaction by deprotonating 2-acetylcyclopentanone and forming hydrogen bonds with the substrates. The origin of enantioselectivity of the reaction is also discussed.
Co-reporter:Wenqi Sun, Jian Wu, Bin Zheng, Yongjun Zhu, Chengbu Liu
Journal of Molecular Structure: THEOCHEM 2007 Volume 809(1–3) pp:161-169
Publication Date(Web):14 May 2007
DOI:10.1016/j.theochem.2007.01.030
The conformational stability, intramolecular and intermolecular H-bond strength, vibrational absorption (VA) and vibrational circular dichroism (VCD) spectra for conformers of (S)-glycidol and their complexes with water have been investigated at B3LYP/6-311+G(d,p) level. The calculated results indicate that the VCD spectra are sensitive to conformational changes of both monomer and complexes. Using the theoretical prediction, we demonstrate that the VCD technique is a powerful approach for determining conformational behavior of chiral molecules.
Co-reporter:Ruoxi Wang, Dongju Zhang, Wenqi Sun, Zhe Han, Chengbu Liu
Journal of Molecular Structure: THEOCHEM 2007 Volume 806(1–3) pp:93-97
Publication Date(Web):31 March 2007
DOI:10.1016/j.theochem.2006.11.012
In order to explore a novel sensor to detect the gaseous molecules, we investigate reactivities of the intrinsic and aluminum-doped (Al-doped) single-walled (8, 0) carbon nanotube (SWCNT) with CO using density functional theory (DFT) calculations. The Al-doped SWCNT presents high sensitivity to CO, compared with the intrinsic SWCNT, as indicated by the calculated geometrical structures and electronic properties for these systems. Al-doped SWCNTs are expected to be a potential candidate for detecting the presence of CO.
The Michael reaction of nitroalkenes catalyzed by a bifunctional-urea is studied using density functional theory (DFT) calculations, to determine the detailed catalytic mechanism and key factors controlling the enantioselectivity. Four reaction channels, corresponding to the different approach modes of nitroalkenes to a chiral scaffold and different processes of second proton transfer, have been characterized. The rate determining step is proton transfer from the amino group of a catalyst to an α-carbon of nitronate, and the enantioselectivity is controlled by the steps involved in carbon–carbon bond formation. The calculated results provide a general model that explains the mechanism and enantioselectivity of the title reaction.The rate determining step of the Michael reaction of nitroalkenes catalyzed by bifunctional-urea is found to be proton transfer from the amino group of the catalyst to the α-carbon of the nitronate, and the enantioselectivity is controlled by the steps involved in carbon–carbon bond formation.
Co-reporter:Ruoxi Wang, Dongju Zhang, Bin Zheng, Zhongnan Qi, Chengbu Liu
Journal of Molecular Structure: THEOCHEM 2006 Volume 772(1–3) pp:13-21
Publication Date(Web):23 October 2006
DOI:10.1016/j.theochem.2006.06.015
The geometrical structures and relative stability of the various possible isomers of silicon–oxygen–sulfur ionic oligomers, (SiOS)n+ and (SiOS)n- (n = 1–6), were studied using quantum chemistry calculations. The ground state of the monomer is a triangular molecule, and that of the dimmer is a rhombic 4MR with two S atoms ending the same Si atom. The most stable isomers for the trimer and tetramer anionic clusters are simply extensions of the rhombic structure, while the tetramer cationic cluster becomes the linear 4MRs with one Si–S defect at the each side. However, for n = 5 and n = 6, the hybrid structures by the 4MRs and hexagonal six-membered ring (6MR) become energetically most favorable configurations. These distinctive structural geometries would provide guide for future experimental detections of these small Si–O–S ionic oligomers.
Chemical Physics Letters 2005 Volume 411(4–6) pp:333-338
Publication Date(Web):15 August 2005
DOI:10.1016/j.cplett.2005.06.055
Abstract
In an effort to search for new inorganic fullerene-like structures, we designed a series of novel silicon–carbon cages, (SiC)n (n = 6–36), based on the uniformly hybrid Si–C four- and six-membered-rings, and researched their geometrical and electronic structures, as well as their relative stabilities using the density function theory. Among these cages, the structures for n = 12, 16, and 36 were found to been energetically more favorable. The calculated disproportionation energy and binding energy per SiC unit show that the (SiC)12 cage is the most stable one among these designed structures. The present calculations not only indicate that silicon–carbon fullerenes are promised to be synthesized in future, but also provide a new way for stabilizing silicon cages by uniformly doping carbon atoms into silicon structures.
Co-reporter:Fancui Meng, Huanjie Wang, Weiren Xu, Chengbu Liu
Chemical Physics 2005 Volume 308(1–2) pp:117-123
Publication Date(Web):10 January 2005
DOI:10.1016/j.chemphys.2004.08.003
Abstract
The geometries of R (R = CH3, CH3O, F, NO2) substituted GC base pair derivatives and their cations have been optimized at B3LYP/6-31G* level and the substituent effects on the neutral and cationic geometric structures and energies have been discussed. The inner reorganization energies of various base pair derivatives and the native GC base pair have been calculated to discuss the substituent effects on the reorganization energy. NBO (natural bond orbital) analysis has been carried out on both the neutral and the cationic systems to investigate the differences of the charge distributions and the electronic structures. The outcomes indicate that 8-CH3O-G:C has the greatest reorganization energy and 8-NO2-G:C has the least, while the other substituted base pairs have a reorganization energy close to that of G:C. The one charge is mostly localized on guanine part after ionization and as high as 0.95e . The bond distances of in the cationic base pair derivatives shortened and that of elongated as compared with the corresponding bond distances of the neutral GC base pair derivatives.
Chemical Physics Letters 2004 Volume 389(4–6) pp:421-426
Publication Date(Web):11 May 2004
DOI:10.1016/j.cplett.2004.03.128
G-tetrad and other 6-thioguanine (SG) incorporated tetrads have been studied in this Letter. The geometries, energies, charge distributions have been discussed. The effects of different cations (K+ and Na+) on the various tetrads have also been studied. The outcomes show that as the SG number increases the tetrad becomes more and more unstable. The Na+ binds more tightly with the tetrad than that of K+ without hydration correction, while considering hydration effects the stability sequence changes to K+ > Na+. Electrostatic potential map of the tetrads have been plotted and the binding sites of cations have been also shown.
Co-reporter:You-Min Sun;Ruo-Xi Wang;Xian-Jie Lin;Shi-Ling Yuan;Cheng-Bu Liu
Chinese Journal of Chemistry 2004 Volume 22(Issue 8) pp:827-830
Publication Date(Web):26 AUG 2010
DOI:10.1002/cjoc.20040220812
The computational results for curcumin at the B3LYP/6-31G(d,p) level show that the enol form of curcumin is more stable than the diketo form because of an intramolecular hydrogen bond, which extends the conjugation effect in the enol chain, formed in the enol structure. Cis-diketone form can not be obtained, presumably due to the strong repulsion between the carbonyl dipoles aligned h parallel. According to the phenolic OH bond dissociation enthalpy, curcumin in its most stable form can be suggested to be a relatively good antioxidant. In order to avoid overcoming H-bond interaction and to improve the antioxidant activity of curcumin, a catechol moiety was incorporated into curcumin for designing a novel antioxidant. It is found that the designed molecule is much more efficient to scavenge radical than curcumin, comparable to vitamin E. Moreover, the ionization potential of the designed molecule is similar to that of curcumin, indicating that the designed molecule can not display the prooxidant effect.
Co-reporter:Dongju Zhang Dr. Dr.;Siwei Bi;Shiling Yuan
Chemistry - A European Journal 2003 Volume 9(Issue 2) pp:
Publication Date(Web):16 JAN 2003
DOI:10.1002/chem.200390051
The reactions of Sc+(3D) with methane, ethane, and propane in the gas phase were studied theoretically by density functional theory. The potential energy surfaces corresponding to [Sc, Cn, H2n+2]+ (n=1–3) were examined in detail at the B3LYP/6-311++G(3df, 3pd)//B3LYP/6-311+G(d,p) level of theory. The performance of this theoretical method was calibrated with respect to the available thermochemical data. Calculations indicated that the reactions of Sc+ with alkanes are multichannel processes which involve two general mechanisms: an addition–elimination mechanism, which is in good agreement with the general mechanism proposed from earlier experiments, and a concerted mechanism, which is presented for the first time in this work. The addition–elimination reactions are favorable at low energy, and the concerted reactions could be alternative pathways at high energy. In most cases, the energetic bottleneck in the addition–elimination mechanism is the initial CC or CH activation. The loss of CH4 and/or C2H6 from Sc++CnH2n+2 (n=2, 3) can proceed along both the initial CC activation branch and the CH activation branch. The loss of H2 from Sc++CnH2n+2 (n=2, 3) can proceed not only by 1,2-H2 and/or 1,3-H2 elimination, but also by 1,1-H2 elimination. The reactivity of Sc+ with alkanes is compared with those reported earlier for the reactions of the late first-row transition-metal ions with alkanes.
New Journal of Chemistry 2002 vol. 26(Issue 3) pp:361-366
Publication Date(Web):14 Feb 2002
DOI:10.1039/B107919K
The models and formalisms for the inner-shell energy barrier of transition metal complexes are considered according to three reorganization criteria. The inner-shell reorganization energies and activation energies of several hexahydrate redox pairs of the first transition row elements, M(H2O)62+/3+
(M=V, Cr, Mn, Fe and Co), are calculated by different formalisms. The extent of the anharmonicities in the vibrations of the metal ion and the ligands in the reorganization process of these complexes is determined. The inner-shell barriers of these redox pairs are dependent on the reorganization model used. The contributions of the individual reactants to the inner-shell barrier are sensitive to the model used.
Co-reporter:Dong-Ju Zhang;Cheng-Bu Liu;Yong-Jun Liu;Hai-Quan Hu
Chinese Journal of Chemistry 2002 Volume 20(Issue 3) pp:
Publication Date(Web):26 AUG 2010
DOI:10.1002/cjoc.20020200304
The mechanism of the reaction of Ni+ (2D) with ethane in the gas-phase was studied by using density functional theory. Both the B3LYP and BLYP functionals with standard all-electron basis sets are used to give the detailed information of the potential energy surface (PES) of [Ni, C2, H6]+. The mechanisms forming the products CH4 and H2 in the reaction of Ni+ with ethane are proposed. The reductive eliminations of CH4 and H2 are typical addition-elimination reactions. Each of the two reactions consists of two elementary steps: C-C or C-H bond activations to form inserted species followed by isomerizations to form product-like intermediate. The rate determining steps for the elimination reactions of forming CH4 and H2 are the isomerizations of the inserted species rather than C-C or C-H bond activations. The elimination reaction of forming H2 was found to be thermodynamically favored compared to that of CH4.
Co-reporter:Dong-Ju Zhang;Cheng-Bu Liu;Hai-Quan Hu;Yong-Jun Liu
Chinese Journal of Chemistry 2001 Volume 19(Issue 10) pp:
Publication Date(Web):26 AUG 2010
DOI:10.1002/cjoc.20010191005
The CF bond activation mechanism of CF3 radical by bare Co+ has been studied by density functional theory. Three local minima and two first-order saddle points were located for the potential energy surface (PES) of [Co, C, F3]+. The activation barrier involving CF bond activation was calculated to be only 14.73 kJ/mol, while the largest barrier of 149.29 kJ/mol on the FES involves CoC bond rupture. The bonding mechanism between Co+, C and F atoms were discussed based on Mulliken population. The relevant bond dissociation energy and thermochemistry data were calculated with the limited experimental values, and the results are in good agreement with the experimental findings.
Chemical Physics Letters 2001 Volume 349(1–2) pp:89-94
Publication Date(Web):23 November 2001
DOI:10.1016/S0009-2614(01)01199-X
A practicable parameter describing magnetic coupling interactions has been suggested. That is the square of overlap integral between the magnetic orbitals in the broken symmetry state. Three molecules, HHeH,[HFH]− and OTi2Cl4, have been calculated with the density functional theory and the broken symmetry approach to inspect this parameter.
Magnetic coupling interactions of a MnIII4 system are investigated by calculations based on density functional theory combined with a broken-symmetry approach (DFT-BS). Three different interactions including ferromagnetic and antiferromagnetic coupling are concomitant in this complex. This magnetic phenomenon of the complex is due to the different bridging angles between the Mn(III) centers in the three different models and the orbital complementarity of the μ-pzbg and μ-OCH3 bridging ligands, which is proven by the analyses of the molecular orbitals. According to the analyses of the magneto-structural correlation, it is revealed that the magnetic coupling interaction switches from ferromagnetic to antiferromagnetic at the point of the bridging angle Mn–(μ-OCH3)–Mn = 99°, which is equal to the value in the origin crystal. Significant correlation between the magnetic properties and the component of the d orbitals in these systems shows that the larger contribution of the dz2 orbital corresponds to the larger ferromagnetic coupling interaction. These results should provide a means to control the magnetic coupling of the polynuclear Mn systems, which is instructive for the design of new molecular magnetic materials.
By employing ab initio quantum mechanical/molecular mechanical (QM/MM) and molecular dynamics (MD) simulations, we have provided further evidence against the previously proposed hydroperoxylation or hydroxylation mechanism of hydroxyethylphosphonate dioxygenase (HEPD). HEPD employs an interesting catalytic cycle based on concatenated bifurcations. The first bifurcation is based on the abstraction of hydrogen atoms from the substrate, which leads to a distal or proximal hydroperoxo species (Fe–OOH or Fe–(OH)O). The second and the third bifurcations refer to the carbon–carbon bond cleavage reaction. And this is achieved through a tridentate intermediate, or employing a proton-shuttle assisted mechanism, in which the residue Glu176 or the FeIVO group serves as a general base. The reaction directions seem to be tunable and show significant environment dependence. This mechanism can provide a comprehensive interpretation for the seemingly contradicting experimental evidences and provide insight into the development of biochemistry and material sciences.







![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-dioctyl-](/data/chemimg/709500/78151-58-3.png)
![Anthra[2,1,9-def:6,5,10-d'e'f']diisoquinoline-1,3,8,10(2H,9H)-tetrone, 2,9-dioctyl-](/data/chemimg/709500/78151-58-3_b.png)