Co-reporter:Akihiko Hatano, Munehiro Okada, Kentaro Dezaki, Seitaro Hirai
Tetrahedron 2015 Volume 71(Issue 7) pp:1095-1100
Publication Date(Web):18 February 2015
DOI:10.1016/j.tet.2014.12.085
We have developed a new route for synthesizing mercapto C-nucleoside possessing a phenyl thiol group, using organometallic reagents. Duplexes incorporating redox-active nucleobase analogues display a high melting temperature under oxidation condition. Originally, we had anticipated the production of mercapto C-nucleoside using a Friedel–Crafts coupling reaction via bis(toluoyl) protected ribose and tert-butyl phenyl sulfide in the presence of Lewis acid. However, an undesired coupling compound was formed by cleavage of the S-tert-butyl group of S-tert-butyl phenyl sulfide by Lewis acids (BF3 Et2O, SnCl4). The highly stereoselective synthesis of mercapto C-nucleoside was, however, achieved by the addition of p-(tert-butyl)thiophenyllithium to a disiloxane-protected 2-deoxyribonolactone. This route showed moderately good yield at all steps. The tert-butyl moiety coupled to the sulfur atom at the phenyl group was converted to a 2-nitrophenylsulfenyl (Nps) group, and the Nps group was easily cleaved by ethanethiol to afford the desired compound and its disulfide dimer.
Co-reporter:Akihiko Hatano, Masayuki Kurosu, Susumu Yonaha, Munehiro Okada and Sanae Uehara
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 40) pp:6900-6905
Publication Date(Web):15 Aug 2013
DOI:10.1039/C3OB41605D
Herein, we describe β-selective coupling between a modified uracil and a deoxyribose to produce functionalized nucleosides catalyzed by thymidine phosphorylase derived from Escherichia coli. This enzyme mediates nucleobase-exchange reactions to convert unnatural nucleosides possessing a large functional group such as a fluorescent molecule, coumarin or pyrene, linked via an alkyl chain at the C5 position of uracil. 5-(Coumarin-7-oxyhex-5-yn)uracil (C4U) displayed 57.2% conversion at 40% DMSO concentration in 1.0 mM phosphate buffer pH 6.8 to transfer thymidine to an unnatural nucleoside with C4U as the base. In the case of using 5-(pyren-1-methyloxyhex-5-yn)uracil (P4U) as the substrate, TP also could catalyse the reaction to generate a product with a very large functional group at 50% DMSO concentration (21.6% conversion). We carried out docking simulations using MF myPrest for the modified uracil bound to the active site of TP. The uracil moiety of the substrate binds to the active site of TP, with the fluorescent moiety linked to the C5 position of the nucleobase located outside the surface of the enzyme. As a consequence, the bulky fluorescent moiety binding to uracil has little influence on the coupling reaction.
Co-reporter:Akihiko Hatano, Munehiro Okada and Gota Kawai
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 36) pp:7327-7333
Publication Date(Web):16 Jul 2012
DOI:10.1039/C2OB25346A
Here, we present the solution structure of a DNA duplex containing a disulfide base pair (S-DNA). The unnatural nucleoside “S” possessing a thiophenyl group as base was incorporated into a self-complementary singled-stranded oligonucleotide. Crosslinking of the disulfide base pair was analyzed by non-denaturing polyacrylamide gel electrophoresis. Under oxidizing conditions a high molecular weight band as 18 mer, corresponding to the double-stranded molecule (5′-GCGASTCGC: 3′-CGCTSAGCG), was found, whereas single-stranded self-complementary 9 mer oligonucleotide GCGASTCGC was detected in the presence of a reducing agent. These results suggest that the oligonucleotide is covalently linked by disulfide bonding under oxidizing conditions, which can be reversibly reduced to two thiol groups under reducing conditions. CD spectrum of S-DNA (CGASTCG) under oxidizing conditions suggested that the duplex had a right-handed double-stranded structure similar to that of natural DNA (B-form, CGATCG). NMR studies confirmed that this CGASTCG resembled natural B-DNA and that the two phenyl rings derived from the disulfide base pairing intercalated into the duplex. However, these two phenyl rings were not positioned in the same plane as the other base pairs. Specifically, NOEs suggest that although CGASTCG adopts a structure similar to B-type DNA, the S-DNA duplex is bent at the point of disulfide base pairing to face the major groove.
Co-reporter:Akihiko Hatano, Yuichi Kanno, Yuya Kondo, Yuta Sunaga, Hatsumi Umezawa, Munehiro Okada, Hideshi Yamada, Ren Iwaki, Atsushi Kato, Koji Fukui
Bioorganic & Medicinal Chemistry (15 January 2017) Volume 25(Issue 2) pp:
Publication Date(Web):15 January 2017
DOI:10.1016/j.bmc.2016.11.053
A series of five new fluorescent deoxynojirimycin (DNJ) conjugates were synthesized and evaluated for their inhibitory effect (IC50) on several α- and β-glucosidases. Three of the conjugates showed enhanced activity. The two synthetic conjugates, DNJ-CF31 and DNJ-Me 2, exhibited improved α-glucosidase inhibitory effects compared to DNJ and miglitol. Interestingly, conjugates 1 and 2 showed strong inhibition of almond-derived β-glucosidase, in contrast to the inhibition tendencies of other inhibitors. Conjugate 5 strongly inhibited rat intestinal maltase, even at 0.10 μM. A docking study indicated that all five conjugates bind to the active site of α-glucosidase (PDB: 3L4V, derived from Homo sapiens). The DNJ portion of the conjugate fits into the cavity of the enzyme, and the fluorescent part locates randomly on the outside surface. Thus, it is likely that these conjugates can specifically recognize intestinal cells, specifically the α-glucosidase on cell membranes.
Co-reporter:Akihiko Hatano, Munehiro Okada and Gota Kawai
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 36) pp:NaN7333-7333
Publication Date(Web):2012/07/16
DOI:10.1039/C2OB25346A
Here, we present the solution structure of a DNA duplex containing a disulfide base pair (S-DNA). The unnatural nucleoside “S” possessing a thiophenyl group as base was incorporated into a self-complementary singled-stranded oligonucleotide. Crosslinking of the disulfide base pair was analyzed by non-denaturing polyacrylamide gel electrophoresis. Under oxidizing conditions a high molecular weight band as 18 mer, corresponding to the double-stranded molecule (5′-GCGASTCGC: 3′-CGCTSAGCG), was found, whereas single-stranded self-complementary 9 mer oligonucleotide GCGASTCGC was detected in the presence of a reducing agent. These results suggest that the oligonucleotide is covalently linked by disulfide bonding under oxidizing conditions, which can be reversibly reduced to two thiol groups under reducing conditions. CD spectrum of S-DNA (CGASTCG) under oxidizing conditions suggested that the duplex had a right-handed double-stranded structure similar to that of natural DNA (B-form, CGATCG). NMR studies confirmed that this CGASTCG resembled natural B-DNA and that the two phenyl rings derived from the disulfide base pairing intercalated into the duplex. However, these two phenyl rings were not positioned in the same plane as the other base pairs. Specifically, NOEs suggest that although CGASTCG adopts a structure similar to B-type DNA, the S-DNA duplex is bent at the point of disulfide base pairing to face the major groove.
Co-reporter:Akihiko Hatano, Masayuki Kurosu, Susumu Yonaha, Munehiro Okada and Sanae Uehara
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 40) pp:NaN6905-6905
Publication Date(Web):2013/08/15
DOI:10.1039/C3OB41605D
Herein, we describe β-selective coupling between a modified uracil and a deoxyribose to produce functionalized nucleosides catalyzed by thymidine phosphorylase derived from Escherichia coli. This enzyme mediates nucleobase-exchange reactions to convert unnatural nucleosides possessing a large functional group such as a fluorescent molecule, coumarin or pyrene, linked via an alkyl chain at the C5 position of uracil. 5-(Coumarin-7-oxyhex-5-yn)uracil (C4U) displayed 57.2% conversion at 40% DMSO concentration in 1.0 mM phosphate buffer pH 6.8 to transfer thymidine to an unnatural nucleoside with C4U as the base. In the case of using 5-(pyren-1-methyloxyhex-5-yn)uracil (P4U) as the substrate, TP also could catalyse the reaction to generate a product with a very large functional group at 50% DMSO concentration (21.6% conversion). We carried out docking simulations using MF myPrest for the modified uracil bound to the active site of TP. The uracil moiety of the substrate binds to the active site of TP, with the fluorescent moiety linked to the C5 position of the nucleobase located outside the surface of the enzyme. As a consequence, the bulky fluorescent moiety binding to uracil has little influence on the coupling reaction.


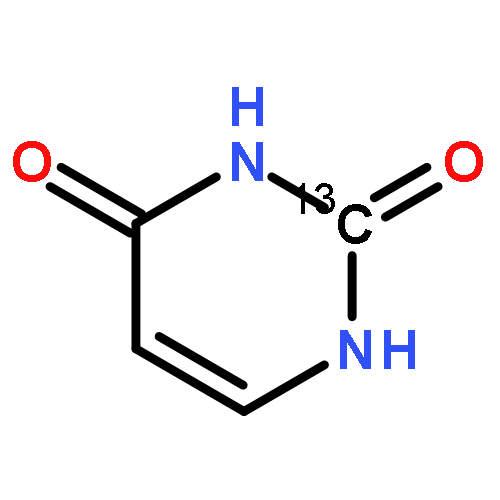
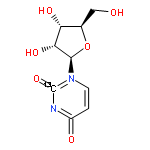
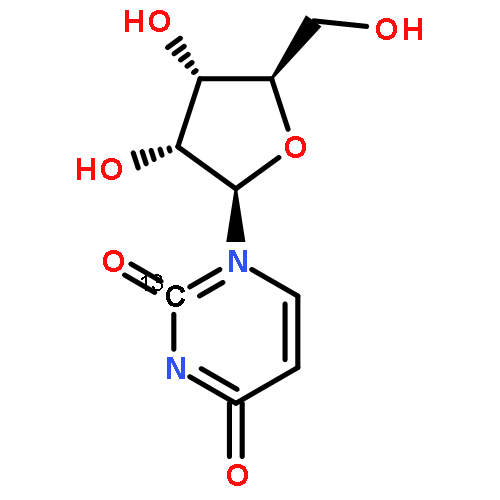
![Benzene,1-bromo-4-[(1,1-dimethylethyl)thio]-](http://img.cochemist.com/ccimg/25800/25752-90-3.png)
![Benzene,1-bromo-4-[(1,1-dimethylethyl)thio]-](http://img.cochemist.com/ccimg/25800/25752-90-3_b.png)
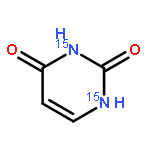
![Benzene,[(1,1-dimethylethyl)thio]-](http://img.cochemist.com/ccimg/3100/3019-19-0.png)
![Benzene,[(1,1-dimethylethyl)thio]-](http://img.cochemist.com/ccimg/3100/3019-19-0_b.png)