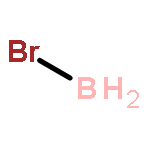
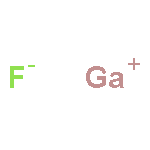Co-reporter:Kayahan Saritas, Tim Mueller, Lucas Wagner, and Jeffrey C. Grossman
Journal of Chemical Theory and Computation May 9, 2017 Volume 13(Issue 5) pp:1943-1943
Publication Date(Web):March 30, 2017
DOI:10.1021/acs.jctc.6b01179
High-throughput calculations based on density functional theory (DFT) methods have been widely implemented in the scientific community. However, depending on both the properties of interest as well as particular chemical/structural phase space, accuracy even for correct trends remains a key challenge for DFT. In this work, we evaluate the use of quantum Monte Carlo (QMC) to calculate material formation energies in a high-throughput environment. We test the performance of automated QMC calculations on 21 compounds with high quality reference data from the Committee on Data for Science and Technology (CODATA) thermodynamic database. We compare our approach to different DFT methods as well as different pseudopotentials, showing that errors in QMC calculations can be progressively improved especially when correct pseudopotentials are used. We determine a set of accurate pseudopotentials in QMC via a systematic investigation of multiple available pseudopotential libraries. We show that using this simple automated recipe, QMC calculations can outperform DFT calculations over a wide set of materials. Out of 21 compounds tested, chemical accuracy has been obtained in formation energies of 11 structures using our QMC recipe, compared to none using DFT calculations.
Co-reporter:Brent D. Keller, Adam Bertuch, J. Provine, Ganesh Sundaram, Nicola Ferralis, and Jeffrey C. Grossman
Chemistry of Materials March 14, 2017 Volume 29(Issue 5) pp:2024-2024
Publication Date(Web):February 27, 2017
DOI:10.1021/acs.chemmater.6b03951
Recent advances in the field of two-dimensional (2D) transition metal dichalcogenide (TMD) materials have indicated that atomic layer deposition (ALD) of the metal oxide and subsequent sulfidation could offer a method for the synthesis of large area two-dimensional materials such as MoS2 with excellent layer control over the entire substrate. However, growing large area oxide films by ALD with sub 1 nm nucleation coalescence remains a significant challenge, and the necessary steps are unexplored. In this work, we demonstrate the necessary process improvements required to achieve sub 1 nm nucleation control by characterization of nucleation domains formed by oxide deposition. Synthesis of the TMD MoS2 from sulfidation of oxide deposited by both thermal ALD from (tBuN)2(NMe2)2Mo and O3 and plasma enhanced ALD (PEALD) from (tBuN)2(NMe2)2Mo and remote O2 plasma was performed. Large uniform MoS2 areas were achieved by optimizing the effects of various growth process conditions and surface treatments on the ALD nucleation and growth of Mo-oxide and the postsulfidation of MoS2. In addition to insights into the control of the oxide deposition, film chemistry analysis during a multistep sulfidation based on less toxic sulfur as compared to H2S was performed for several temperature profiles revealing sulfur incorporation and molybdenum reduction at low temperatures but higher temperatures required for 2H crystal structure formation. The knowledge gained of the ALD, PEALD, and postsulfidation was leveraged to demonstrate tunable film thickness and centimeter-scale monolayer growth. Material quality can be studied independently of the MoS2 layer count as demonstrated by the control of the monolayer photoluminescence intensity by the temperature ramp rate during sulfidation.
Co-reporter:Kayahan Saritas and Jeffrey C. Grossman
The Journal of Physical Chemistry C December 7, 2017 Volume 121(Issue 48) pp:26677-26677
Publication Date(Web):November 8, 2017
DOI:10.1021/acs.jpcc.7b09437
We investigate the isomerization enthalpy of the dihydroazulene/vinylheptafulvene (DHA/VHF) molecular photoswitch system derivatives using electronic structure calculation methods including density functional theory (DFT), quantum Monte Carlo (QMC), and coupled cluster (CCSD(T)). Recent efforts have focused on tuning the isomerization enthalpy of the photoswitch for solar thermal energy storage applications using substitutional functional groups on its five- and seven-membered carbon rings, predominantly using DFT for the energy predictions. However, using the higher accuracy QMC and CCSD(T) methods, we show that in many cases DFT incorrectly predicts the isomerization enthalpy, and the errors depend on the functional groups substituted and the choice of the DFT functional. Isomerization of the DHA to VHF molecule is an electrocyclic ring-opening reaction on the five-membered ring of the DHA isomer. We find that the DFT errors are correlated to the electrocyclic ring-opening reactions of cyclobutene and cyclo-1,3-hexadiene, such that the DFT error changes monotonically with the size of the carbon ring, although QMC and CCSD(T) results are in a good agreement irrespective of the ring size. Using the QMC and CCSD(T) isomerization enthalpies, we predict gravimetric energy densities of the DHA derivatives for solar thermal storage applications. Our results show that suitable substitutions on DHA can yield gravimetric storage densities as large as 732 kJ/kg.
Co-reporter:Jatin J. Patil;Brendan D. Smith
RSC Advances (2011-Present) 2017 vol. 7(Issue 19) pp:11537-11542
Publication Date(Web):2017/02/13
DOI:10.1039/C7RA00562H
Nanoporous silicon (NPSi) has drawn recent interest because of its potential in a range of applications such as battery anodes, photocatalysis, thermoelectrics, and filtration membranes. However, the inexpensive and scalable manufacturing of high aspect ratio porous structures on the nanometer scale has been difficult due to the reliance of current methods on complex and expensive equipment used for techniques such as anodization or photolithography. Here, we report a method of producing NPSi with sub-10 nm pore sizes and aspect ratios as high as 400 : 1 by leveraging the nucleation of sputtered noble metals on the Si surface, followed by metal-assisted chemical etching (MACE). The technique is capable of producing NPSi in an intrinsically scalable manner. Samples are characterized with SEM and TEM, along with vertical and horizontal FIB cross-sectional milling to elucidate the porous structure at several μm of depth within the substrate. Following preparation of the NPSi, it is functionalized with Al2O3 and TiO2 via atomic layer deposition (ALD). TiO2-functionalized NPSi exhibits reflectivity of 6–8% for visible wavelengths, and 2–3% in the infrared – showing its promise as a robust and functional porous substrate. The developed approach of employing MACE with sputtered nucleated catalysts facilitates the scalable fabrication of functional ultra-high aspect-ratio nanopores in silicon.
Co-reporter:Huashan Li
Advanced Science 2017 Volume 4(Issue 8) pp:
Publication Date(Web):2017/08/01
DOI:10.1002/advs.201600467
Control of both the regularity of a material ensemble and nanoscale architecture provides unique opportunities to develop novel thermoelectric applications based on 2D materials. As an example, the authors explore the electronic and thermal properties of functionalized graphene nanoribbons (GNRs) in the single-sheet and helical architectures using multiscale simulations. The results suggest that appropriate functionalization enables precise tuning of the doping density in a planar donor/acceptor GNR ensemble without the need to introduce an explicit dopant, which is critical to the optimization of power factor. In addition, the self-interaction between turns of a GNR may induce long-range disorder along the helical axis, which suppresses the thermal contribution from phonons with long wavelengths, leading to anomalous length independent phonon thermal transport in the quasi-1D system.
Co-reporter:Grace G. D. Han;Kun-Hua Tu;Farnaz Niroui;Wenshuo Xu;Si Zhou;Xiaochen Wang;Vladimir Bulović;Caroline A. Ross;Jamie H. Warner
Advanced Functional Materials 2017 Volume 27(Issue 45) pp:
Publication Date(Web):2017/12/01
DOI:10.1002/adfm.201703688
AbstractMonolayer 2D MoS2 grown by chemical vapor deposition is nanopatterned into nanodots, nanorods, and hexagonal nanomesh using block copolymer (BCP) lithography. The detailed atomic structure and nanoscale geometry of the nanopatterned MoS2 show features down to 4 nm with nonfaceted etching profiles defined by the BCP mask. Atomic resolution annular dark field scanning transmission electron microscopy reveals the nanopatterned MoS2 has minimal large-scale crystalline defects and enables the edge density to be measured for each nanoscale pattern geometry. Photoluminescence spectroscopy of nanodots, nanorods, and nanomesh areas shows strain-dependent spectral shifts up to 15 nm, as well as reduction in the PL efficiency as the edge density increases. Raman spectroscopy shows mode stiffening, confirming the release of strain when it is nanopatterned by BCP lithography. These results show that small nanodots (≈19 nm) of MoS2 2D monolayers still exhibit strong direct band gap photoluminescence (PL), but have PL quenching compared to pristine material from the edge states. This information provides important insights into the structure–PL property correlations of sub-20 nm MoS2 structures that have potential in future applications of 2D electronics, optoelectronics, and photonics.
Co-reporter:Neelkanth M. BardhanPriyank V. Kumar, Zeyang Li, Hidde L. Ploegh, Jeffrey C. GrossmanAngela M. Belcher, Guan-Yu Chen
ACS Nano 2017 Volume 11(Issue 2) pp:
Publication Date(Web):January 13, 2017
DOI:10.1021/acsnano.6b06979
With the global rise in incidence of cancer and infectious diseases, there is a need for the development of techniques to diagnose, treat, and monitor these conditions. The ability to efficiently capture and isolate cells and other biomolecules from peripheral whole blood for downstream analyses is a necessary requirement. Graphene oxide (GO) is an attractive template nanomaterial for such biosensing applications. Favorable properties include its two-dimensional architecture and wide range of functionalization chemistries, offering significant potential to tailor affinity toward aromatic functional groups expressed in biomolecules of interest. However, a limitation of current techniques is that as-synthesized GO nanosheets are used directly in sensing applications, and the benefits of their structural modification on the device performance have remained unexplored. Here, we report a microfluidic-free, sensitive, planar device on treated GO substrates to enable quick and efficient capture of Class-II MHC-positive cells from murine whole blood. We achieve this by using a mild thermal annealing treatment on the GO substrates, which drives a phase transformation through oxygen clustering. Using a combination of experimental observations and MD simulations, we demonstrate that this process leads to improved reactivity and density of functionalization of cell capture agents, resulting in an enhanced cell capture efficiency of 92 ± 7% at room temperature, almost double the efficiency afforded by devices made using as-synthesized GO (54 ± 3%). Our work highlights a scalable, cost-effective, general approach to improve the functionalization of GO, which creates diverse opportunities for various next-generation device applications.Keywords: enhanced cell capture efficiency; graphene oxide; improved functionalization; microfluidic-free; oxygen clustering; phase transformation; thermal annealing;
Co-reporter:Marco Bernardi and Jeffrey C. Grossman
Energy & Environmental Science 2016 vol. 9(Issue 7) pp:2197-2218
Publication Date(Web):05 May 2016
DOI:10.1039/C6EE01010E
Photovoltaic (PV) solar cells convert solar energy to electricity through a cascade of microscopic processes spanning over 10 order of magnitudes of time and length. PV conversion involves a complex interplay of photons, charge carriers, and excited states. Processes following light absorption include generation of charge carriers or excitons, exciton dissociation over nanometer lengths and subpicosecond times, and carrier transport over ns–ms times and nm–mm lengths. Computer calculations have become an indispensable tool to understand and engineer solar cells across length and time scales. In this article, we examine the microscopic processes underlying PV conversion and review state-of-the-art computational methods to study PV solar cells. Recent developments and future research challenges are outlined.
Co-reporter:David Zhitomirsky;Eugene Cho
Advanced Energy Materials 2016 Volume 6( Issue 6) pp:
Publication Date(Web):
DOI:10.1002/aenm.201502006
Closed cycle systems offer an opportunity for solar energy harvesting and storage all within the same material. Photon energy is stored within the chemical conformations of molecules and is retrieved by a triggered release in the form of heat. Until now, such solar thermal fuels (STFs) have been largely unavailable in the solid-state, which would enable them to be utilized for a multitude of applications. A polymer STF storage platform is synthesized employing STFs in the solid-state. This approach enables uniform films capable of appreciable heat storage of up to 30 Wh kg−1 and that can withstand temperature of up to 180 °C. For the first time a macroscopic energy release is demonstrated using spatial infrared heat maps with up to a 10 °C temperature change. These findings pave the way for developing highly efficient and high energy density STFs for applications in the solid-state.
Co-reporter:Beibei Xu, Huashan Li, Haoqi Li, Andrew J. Wilson, Lin Zhang, Ke Chen, Katherine A. Willets, Fei Ren, Jeffrey C. Grossman, and Shenqiang Ren
Nano Letters 2016 Volume 16(Issue 4) pp:2851-2859
Publication Date(Web):March 21, 2016
DOI:10.1021/acs.nanolett.6b00712
Organic charge-transfer superstructures are enabling new interfacial electronics, such as organic thermoelectrics, spin-charge converters, and solar cells. These carbon-based materials could also play an important role in spin-based electronics due to their exceptionally long spin lifetime. However, to explore these potentials a coherent design strategy to control interfacial charge-transfer interaction is indispensable. Here we report that the control of organic crystallization and interfacial electron coupling are keys to dictate external stimuli responsive behaviors in organic charge-transfer superstructures. The integrated experimental and computational study reveals the importance of chemically driven interfacial coupling in organic charge-transfer superstructures. Such degree of engineering opens up a new route to develop a new generation of functional charge-transfer materials, enabling important advance in all organic interfacial electronics.
Co-reporter:David Cohen-Tanugi, Li-Chiang Lin, and Jeffrey C. Grossman
Nano Letters 2016 Volume 16(Issue 2) pp:1027-1033
Publication Date(Web):January 25, 2016
DOI:10.1021/acs.nanolett.5b04089
While single-layer nanoporous graphene (NPG) has shown promise as a reverse osmosis (RO) desalination membrane, multilayer graphene membranes can be synthesized more economically than the single-layer material. In this work, we build upon the knowledge gained to date toward single-layer graphene to explore how multilayer NPG might serve as a RO membrane in water desalination using classical molecular dynamic simulations. We show that, while multilayer NPG exhibits similarly promising desalination properties to single-layer membranes, their separation performance can be designed by manipulating various configurational variables in the multilayer case. This work establishes an atomic-level understanding of the effects of additional NPG layers, layer separation, and pore alignment on desalination performance, providing useful guidelines for the design of multilayer NPG membranes.
Co-reporter:Brent D. Keller, Nicola Ferralis, and Jeffrey C. Grossman
Nano Letters 2016 Volume 16(Issue 5) pp:2951-2957
Publication Date(Web):March 31, 2016
DOI:10.1021/acs.nanolett.5b04735
Disordered carbon materials, both amorphous and with long-range order, have been used in a variety of applications, from conductive additives and contact materials to transistors and photovoltaics. Here we show a flexible solution-based method of preparing thin films with tunable electrical properties from suspensions of ball-milled coals following centrifugation. The as-prepared films retain the rich carbon chemistry of the starting coals with conductivities ranging over orders of magnitude, and thermal treatment of the resulting films further tunes the electrical conductivity in excess of 7 orders of magnitude. Optical absorption measurements demonstrate tunable optical gaps from 0 to 1.8 eV. Through low-temperature conductivity measurements and Raman spectroscopy, we demonstrate that variable range hopping controls the electrical properties in as-prepared and thermally treated films and that annealing increases the sp2 content, localization length, and disorder. The measured hopping energies demonstrate electronic properties similar to amorphous carbon materials and reduced graphene oxide. Finally, Joule heating devices were fabricated from coal-based films, and temperatures as high as 285 °C with excellent stability were achieved.
Co-reporter:Jeong Yun Kim and Jeffrey C. Grossman
Nano Letters 2016 Volume 16(Issue 7) pp:4203-4209
Publication Date(Web):June 20, 2016
DOI:10.1021/acs.nanolett.6b01073
Crystalline C60 is an appealing candidate material for thermoelectric (TE) applications due to its extremely low thermal conductivity and potentially high electrical conductivity with metal atom intercalation. We investigate the TE properties of crystalline C60 intercalated with alkali and alkaline earth metals using both classical and quantum mechanical calculations. For the electronic structure, our results show that variation of intercalated metal atoms has a large impact on energy dispersions, which leads to broad tunability of the power factor. For the thermal transport, we show that dopants introduce strong phonon scattering into crystalline C60, leading to considerably lower thermal conductivity. Taking both into account, our calculations suggest that appropriate choice of metal atom intercalation in crystalline C60 could yield figures of merit near 1 at room temperature.
Co-reporter:Sangjin Lee;David Zhitomirsky
Advanced Functional Materials 2016 Volume 26( Issue 10) pp:1554-1562
Publication Date(Web):
DOI:10.1002/adfm.201504816
A realistic CQD solid model is developed that computes the charge carrier mobility using hopping transport models within an ensemble of individual CQD units. Large decreases in electron mobility of up to 70% as compared to the monodisperse case are observed when the energetic disorder in CQD films lies in the typical experimental range of 10%–15%. Furthermore, it is suggested that tailored and potentially experimentally achievable re-arrangement of the CQD size ensemble combined with spatial doping control can mediate the reduction in mobility even in highly dispersive cases, and presents an avenue toward improved mobility and photovoltaic performance by up to 9% by leveraging fast carrier transport channels in highly polydisperse materials.
Co-reporter:Huashan Li, David Zhitomirsky, and Jeffrey C. Grossman
Chemistry of Materials 2016 Volume 28(Issue 6) pp:1888
Publication Date(Web):February 21, 2016
DOI:10.1021/acs.chemmater.6b00167
PbS nanoplatelets (NPLs) are proposed as robust materials for novel optoelectronic devices. Compared to quantum dot assemblies, ab initio simulations are employed to show that such pseudo-two-dimensional systems may provide stronger absorption and higher carrier mobility due to the distinct wave function distributions, large electronic couplings, and small hopping barriers. More importantly, both energetic and spatial traps are absent in conditions far from charge balance, indicating an extraordinary robustness against off-stoichiometry as a result of surface homogeneity and sufficient cross-linking. Based on our findings, we present several types of optoelectronic device architectures spanning photovoltaics and photodetectors that could take advantage of the superior properties found in NPLs.
Co-reporter:Brendan D. Smith, Jatin J. Patil, Nicola Ferralis, and Jeffrey C. Grossman
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 12) pp:8043
Publication Date(Web):March 21, 2016
DOI:10.1021/acsami.6b01927
Nanoporous silicon (NPSi) has received significant attention for its potential to contribute to a large number of applications, but has not yet been extensively implemented because of the inability of current state-of-the-art nanofabrication techniques to achieve sufficiently small pore size, high aspect ratio, and process scalability. In this work we describe the fabrication of NPSi via a modified metal-assisted chemical etching (MACE) process in which silica-shell gold nanoparticle (SiO2–AuNP) monolayers self-assemble from solution onto a silicon substrate. Exposure to the MACE etchant solution results in the rapid consumption of the SiO2 spacer shell, leaving well-spaced arrays of bare AuNPs on the substrate surface. Particles then begin to catalyze the etching of nanopore arrays without interruption, resulting in the formation of highly anisotropic individual pores. The excellent directionality of pore formation is thought to be promoted by the homogeneous interparticle spacing of the gold core nanocatalysts, which allow for even hole injection and subsequent etching along preferred crystallographic orientations. Electron microscopy and image analysis confirm the ability of the developed technique to produce micrometer-scale arrays of sub 10 nm nanopores with narrow size distributions and aspect ratios of over 100:1. By introducing a scalable process for obtaining high aspect ratio pores in a novel size regime, this work opens the door to implementation of NPSi in numerous devices and applications.Keywords: metal-assisted chemical etching; nanoporous silicon; self-assembly; silica-shell gold nanoparticles; ultrahigh aspect ratio
Co-reporter:David Zhitomirsky and Jeffrey C. Grossman
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 39) pp:26319
Publication Date(Web):September 9, 2016
DOI:10.1021/acsami.6b08034
There is tremendous growth in fields where small functional molecules and colloidal nanomaterials are integrated into thin films for solid-state device applications. Many of these materials are synthesized in solution and there often exists a significant barrier to transition them into the solid state in an efficient manner. Here, we develop a methodology employing an electrodepositable copolymer consisting of small functional molecules for applications in solar energy harvesting and storage. We employ azobenzene solar thermal fuel polymers and functionalize them to enable deposition from low concentration solutions in methanol, resulting in uniform and large-area thin films. This approach enables conformal deposition on a variety of conducting substrates that can be either flat or structured depending on the application. Our approach further enables control over film growth via electrodepsition conditions and results in highly uniform films of hundreds of nanometers to microns in thickness. We demonstrate that this method enables superior retention of solar thermal fuel properties, with energy densities of ∼90 J/g, chargeability in the solid state, and exceptional materials utilization compared to other solid-state processing approaches. This novel approach is applicable to systems such as photon upconversion, photovoltaics, photosensing, light emission, and beyond, where small functional molecules enable solid-state applications.Keywords: azobenzene; electrodeposition; polymers; solar thermal fuels; thin-films
Co-reporter:Eric Johlin, Ahmed Al-Obeidi, Gizem Nogay, Michael Stuckelberger, Tonio Buonassisi, and Jeffrey C. Grossman
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 24) pp:15169-15176
Publication Date(Web):May 26, 2016
DOI:10.1021/acsami.6b00033
While low hole mobilities limit the current collection and efficiency of hydrogenated amorphous silicon (a-Si:H) photovoltaic devices, attempts to improve mobility of the material directly have stagnated. Herein, we explore a method of utilizing nanostructuring of a-Si:H devices to allow for improved hole collection in thick absorber layers. This is achieved by etching an array of 150 nm diameter holes into intrinsic a-Si:H and then coating the structured material with p-type a-Si:H and a conformal zinc oxide transparent conducting layer. The inclusion of these nanoholes yields relative power conversion efficiency (PCE) increases of ∼45%, from 7.2 to 10.4% PCE for small area devices. Comparisons of optical properties, time-of-flight mobility measurements, and internal quantum efficiency spectra indicate this efficiency is indeed likely occurring from an improved collection pathway provided by the nanostructuring of the devices. Finally, we estimate that through modest optimizations of the design and fabrication, PCEs of beyond 13% should be obtainable for similar devices.
Co-reporter:Shreya H. Dave, Chuncheng Gong, Alex W. Robertson, Jamie H. Warner, and Jeffrey C. Grossman
ACS Nano 2016 Volume 10(Issue 8) pp:7515
Publication Date(Web):July 10, 2016
DOI:10.1021/acsnano.6b02391
Graphene oxide (GO) and reduced GO (rGO) are the only variants of graphene that can be manufactured at the kilogram scale, and yet the widely accepted model for their structure has largely relied on indirect evidence. Notably, existing high-resolution transmission electron microscopy (HRTEM) studies of graphene oxide report long-range order of sp2 lattice with isolated defect clusters. Here, we present HRTEM evidence of a different structural form of GO, where nanocrystalline regions of sp2 lattice are surrounded by regions of disorder. The presence of contaminants that adsorb to the surface of the material at room temperature normally prevents direct observation of the intrinsic atomic structure of this defective GO. To overcome this, we use an in situ heating holder within an aberration-corrected TEM (AC-TEM) to study the atomic structure of this nanocrystalline graphene oxide from room temperature to 700 °C. As the temperature increases to above 500 °C, the adsorbates detach from the GO and the underlying atomic structure is imaged to be small 2–4 nm crystalline domains within a polycrystalline GO film. By combining spectroscopic evidence with the AC-TEM data, we support the dynamic interpretation of the structural evolution of graphene oxide.Keywords: AC-TEM; graphene oxide; surface contaminants; TEM
Co-reporter:Huashan Li, David Zhitomirsky, Shreya Dave, and Jeffrey C. Grossman
ACS Nano 2016 Volume 10(Issue 1) pp:606
Publication Date(Web):January 8, 2016
DOI:10.1021/acsnano.5b05626
Colloidal quantum dots (CQDs) are highly versatile nanoscale optoelectronic building blocks, but despite their materials engineering flexibility, there is a considerable lack of fundamental understanding of their electronic structure as they couple within thin films. By employing a joint experimental–theoretical study, we reveal the impact of connectivity in CQD assemblies, going beyond the single CQD picture. High-resolution transmission electron microscopy (HR-TEM) demonstrates connectivity motifs across different CQD sizes and length scales and provides the necessary perspective to build robust computational models to systematically study the achievable degree of connectivity in these materials. We focused on state-of-the-art surface ligand treatments, taking into account both the degree of connectivity and nanocrystal orientation, and performed ab initio simulations within the phonon-assisted hopping regime. Importantly, both the TEM studies and our simulation results revealed morphological and electronic defects that could dramatically reduce optoelectronic performance, and yet would not have been captured within a single CQD model that neglects connectivity. We calculate carrier mobility in the presence of such defect states and conclude that the best-achievable CQD assemblies for optoelectronics will require a modest degree of fusing via the {001} facet, followed by atomic ligand passivation to generate a clean band gap and unprecedentedly high charge transport.Keywords: charge transport; colloidal QD; density functional theory; high-resolution TEM; necking;
Co-reporter:Yun Liu, Nicola Ferralis, L. Taras Bryndzia, Jeffrey C. Grossman
Carbon 2016 101() pp: 361-367
Publication Date(Web):May 2016
DOI:10.1016/j.carbon.2016.02.017
Rapid, non-destructive characterization of molecular level chemistry for organic matter (OM) is experimentally challenging. Raman spectroscopy is one of the most widely used techniques for non-destructive chemical characterization, although it currently does not provide detailed identification of molecular components in OM, due to the combination of diffraction-limited spatial resolution and poor applicability of peak-fitting algorithms. Here, we develop a genome-inspired collective molecular structure fingerprinting approach, which utilizes ab initio calculations and data mining techniques to extract molecular level chemistry from the Raman spectra of OM. We illustrate the power of such an approach by identifying representative molecular fingerprints in OM, for which the molecular chemistry is to date inaccessible using non-destructive characterization techniques. Chemical properties such as aromatic cluster size distribution and H/C ratio can now be quantified directly using the identified molecular fingerprints. Our approach will enable non-destructive identification of chemical signatures with their correlation to the preservation of biosignatures in OM, accurate detection and quantification of environmental contamination, as well as objective assessment of OM with respect to their chemical contents.
Co-reporter:Jeong Yun Kim and Jeffrey C. Grossman
Nano Letters 2015 Volume 15(Issue 5) pp:2830-2835
Publication Date(Web):April 6, 2015
DOI:10.1021/nl504257q
Graphene superlattices made with chemical functionalization offer the possibility of tuning both the thermal and electronic properties via nanopatterning of the graphene surface. Using classical and quantum mechanical calculations, we predict that suitable chemical functionalization of graphene can introduce peaks in the density of states at the band edge that result in a large enhancement in the Seebeck coefficient, leading to an increase in the room-temperature power factor of a factor of 2 compared to pristine graphene, despite the degraded electrical conductivity. Furthermore, the presence of patterns on graphene reduces the thermal conductivity, which when taken together leads to an increase in the figure of merit for functionalized graphene by up to 2 orders of magnitude over that of pristine graphene, reaching its maximum ZT ∼ 3 at room temperature according to our calculations. These results suggest that appropriate chemical functionalization could lead to efficient graphene-based thermoelectric materials.
Co-reporter:Maurizia Palummo, Marco Bernardi, and Jeffrey C. Grossman
Nano Letters 2015 Volume 15(Issue 5) pp:2794-2800
Publication Date(Web):March 23, 2015
DOI:10.1021/nl503799t
Light emission in two-dimensional (2D) transition metal dichalcogenides (TMDs) changes significantly with the number of layers and stacking sequence. While the electronic structure and optical absorption are well understood in 2D-TMDs, much less is known about exciton dynamics and radiative recombination. Here, we show first-principles calculations of intrinsic exciton radiative lifetimes at low temperature (4 K) and room temperature (300 K) in TMD monolayers with the chemical formula MX2 (X = Mo, W, and X = S, Se), as well as in bilayer and bulk MoS2 and in two MX2 heterobilayers. Our results elucidate the time scale and microscopic origin of light emission in TMDs. We find radiative lifetimes of a few picoseconds at low temperature and a few nanoseconds at room temperature in the monolayers and slower radiative recombination in bulk and bilayer than in monolayer MoS2. The MoS2/WS2 and MoSe2/WSe2 heterobilayers exhibit very long-lived (∼20–30 ns at room temperature) interlayer excitons constituted by electrons localized on the Mo-based and holes on the W-based monolayer. The wide radiative lifetime tunability, together with the ability shown here to predict radiative lifetimes from computations, hold unique potential to manipulate excitons in TMDs and their heterostructures for application in optoelectronics and solar energy conversion.
Co-reporter:Huashan Li;David A. Strubbe
Advanced Functional Materials 2015 Volume 25( Issue 32) pp:5199-5205
Publication Date(Web):
DOI:10.1002/adfm.201501906
In-plane heterostructure engineering provides unique opportunities to control device properties. Here, a single-sheet solar cell made of a graphene sheet functionalized into 1D channels is explored. Compared to vertical heterostructure architectures based on 2D materials, the single-sheet solar cell shows potential for improved robustness against defects, enhancement of polaron dissociation, extra freedom for functionalization, and coverage of the entire solar spectrum. The partition width, device length, and functionalizations can be tuned independently to optimize the key optoelectronic properties for photovoltaic performance.
Co-reporter:Li-Chiang Lin, Jongwon Choi and Jeffrey C. Grossman
Chemical Communications 2015 vol. 51(Issue 80) pp:14921-14924
Publication Date(Web):14 Aug 2015
DOI:10.1039/C5CC05969K
We computationally demonstrate that two-dimensional covalent triazine frameworks (CTFs) provide opportunities in water desalination. By varying the chemical building blocks, the pore structure, chemistry, and membrane performance can be designed, leading to two orders of magnitude higher water permeability than polyamide membranes while maintaining excellent ability to reject salts.
Co-reporter:Kayahan Saritas
The Journal of Physical Chemistry C 2015 Volume 119(Issue 9) pp:5074-5079
Publication Date(Web):January 30, 2015
DOI:10.1021/jp510597e
Co-reporter:David Cohen-Tanugi, Ronan K. McGovern, Shreya H. Dave, John H. Lienhard and Jeffrey C. Grossman
Energy & Environmental Science 2014 vol. 7(Issue 3) pp:1134-1141
Publication Date(Web):04 Feb 2014
DOI:10.1039/C3EE43221A
In the face of growing water scarcity, it is critical to understand the potential of saltwater desalination as a long-term water supply option. Recent studies have highlighted the promise of new membrane materials that could desalinate water while exhibiting far greater permeability than conventional reverse osmosis (RO) membranes, but the question remains whether higher permeability can translate into significant reductions in the cost of desalinating water. Here, we address a critical question by evaluating the potential of such ultra-permeable membranes (UPMs) to improve the performance and cost of RO. By modeling the mass transport inside RO pressure vessels, we quantify how much a tripling in the water permeability of a membrane would reduce the energy consumption or the number of required pressure vessels for a given RO plant. We find that a tripling in permeability would allow for 44% fewer pressure vessels or 15% less energy for a seawater RO plant with a given capacity and recovery ratio. Moreover, a tripling in permeability would result in 63% fewer pressure vessels or 46% less energy for brackish water RO. However, we also find that the energy savings of UPMs exhibit a law of diminishing returns due to thermodynamics and concentration polarization at the membrane surface.
Co-reporter:David Cohen-Tanugi and Jeffrey C. Grossman
Nano Letters 2014 Volume 14(Issue 11) pp:6171-6178
Publication Date(Web):October 30, 2014
DOI:10.1021/nl502399y
Recent advances in the development of nanoporous graphene (NPG) hold promise for the future of water supply by reverse osmosis (RO) desalination. But while previous studies have highlighted the potential of NPG as an RO membrane, there is less understanding as to whether NPG is strong enough to maintain its mechanical integrity under the high hydraulic pressures inherent to the RO desalination process. Here, we show that an NPG membrane can maintain its mechanical integrity in RO but that the choice of substrate for graphene is critical to this performance. Using molecular dynamics simulations and continuum fracture mechanics, we show that an appropriate substrate with openings smaller than 1 μm would allow NPG to withstand pressures exceeding 57 MPa (570 bar) or ten times more than typical pressures for seawater RO. Furthermore, we demonstrate that NPG membranes exhibit an unusual mechanical behavior in which greater porosity may help the membrane withstand even higher pressures.
Co-reporter:Yun Liu and Jeffrey C. Grossman
Nano Letters 2014 Volume 14(Issue 12) pp:7046-7050
Publication Date(Web):November 5, 2014
DOI:10.1021/nl5034073
Solar thermal fuels (STF) store the energy of sunlight, which can then be released later in the form of heat, offering an emission-free and renewable solution for both solar energy conversion and storage. However, this approach is currently limited by the lack of low-cost materials with high energy density and high stability. In this Letter, we present an ab initio high-throughput computational approach to accelerate the design process and allow for searches over a broad class of materials. The high-throughput screening platform we have developed can run through large numbers of molecules composed of earth-abundant elements and identifies possible metastable structures of a given material. Corresponding isomerization enthalpies associated with the metastable structures are then computed. Using this high-throughput simulation approach, we have discovered molecular structures with high isomerization enthalpies that have the potential to be new candidates for high-energy density STF. We have also discovered physical principles to guide further STF materials design through structural analysis. More broadly, our results illustrate the potential of using high-throughput ab initio simulations to design materials that undergo targeted structural transitions.
Co-reporter:Rajamani Raghunathan, Eric Johlin, and Jeffrey C. Grossman
Nano Letters 2014 Volume 14(Issue 9) pp:4943-4950
Publication Date(Web):June 25, 2014
DOI:10.1021/nl501020q
In photovoltaic devices, the bulk disorder introduced by grain boundaries (GBs) in polycrystalline silicon is generally considered to be detrimental to the physical stability and electronic transport of the bulk material. However, at the extremum of disorder, amorphous silicon is known to have a beneficially increased band gap and enhanced optical absorption. This study is focused on understanding and utilizing the nature of the most commonly encountered Σ3 GBs, in an attempt to balance incorporation of the advantageous properties of amorphous silicon while avoiding the degraded electronic transport of a fully amorphous system. A combination of theoretical methods is employed to understand the impact of ordered Σ3 GBs on the material properties and full-device photovoltaic performance.
Co-reporter:Patrick R. Brown, Donghun Kim, Richard R. Lunt, Ni Zhao, Moungi G. Bawendi, Jeffrey C. Grossman, and Vladimir Bulović
ACS Nano 2014 Volume 8(Issue 6) pp:5863
Publication Date(Web):May 13, 2014
DOI:10.1021/nn500897c
The electronic properties of colloidal quantum dots (QDs) are critically dependent on both QD size and surface chemistry. Modification of quantum confinement provides control of the QD bandgap, while ligand-induced surface dipoles present a hitherto underutilized means of control over the absolute energy levels of QDs within electronic devices. Here, we show that the energy levels of lead sulfide QDs, measured by ultraviolet photoelectron spectroscopy, shift by up to 0.9 eV between different chemical ligand treatments. The directions of these energy shifts match the results of atomistic density functional theory simulations and scale with the ligand dipole moment. Trends in the performance of photovoltaic devices employing ligand-modified QD films are consistent with the measured energy level shifts. These results identify surface-chemistry-mediated energy level shifts as a means of predictably controlling the electronic properties of colloidal QD films and as a versatile adjustable parameter in the performance optimization of QD optoelectronic devices.Keywords: density functional theory; lead sulfide; ligands; nanocrystals; photovoltaics; quantum dots; solar cells; ultraviolet photoelectron spectroscopy
Co-reporter:E. Durgun ; H. Manzano ; P. V. Kumar
The Journal of Physical Chemistry C 2014 Volume 118(Issue 28) pp:15214-15219
Publication Date(Web):July 3, 2014
DOI:10.1021/jp408325f
Calcium silicate compounds belong to a complicated class of silicates. Among their many industrial applications, calcium silicates are heavily used as a building material as they constitute the main ingredient in today’s cement clinker. We report here an extensive surface analysis of synthetic calcium silicate phases (tricalcium silicate, C3S, and dicalcium silicate, C2S) using first-principles computational methods. We calculate surface energies (γ) for all lower-index orientations and determine the most stable surfaces as well as the equilibrium Wulff structures. We analyze the variation of γ with surface coordination number and find an interesting and unexpected trend where loss of coordination of ionic Ca and O atoms can lower γ. The stability of surface orientations is examined as a function of oxygen partial pressure. Finally, we compute the energy required to remove Ca from different surfaces and find that it is inversely proportional to γ, supporting the energetic preference of extracting atoms from higher energy surfaces. Knowledge of the atomic structure and properties of calcium silicate surfaces is important for understanding and controlling the hydration of such systems.
Co-reporter:Marco Bernardi, Maurizia Palummo, and Jeffrey C. Grossman
Nano Letters 2013 Volume 13(Issue 8) pp:3664-3670
Publication Date(Web):June 10, 2013
DOI:10.1021/nl401544y
Graphene and monolayer transition metal dichalcogenides (TMDs) are promising materials for next-generation ultrathin optoelectronic devices. Although visually transparent, graphene is an excellent sunlight absorber, achieving 2.3% visible light absorbance in just 3.3 Å thickness. TMD monolayers also hold potential as sunlight absorbers, and may enable ultrathin photovoltaic (PV) devices due to their semiconducting character. In this work, we show that the three TMD monolayers MoS2, MoSe2, and WS2 can absorb up to 5–10% incident sunlight in a thickness of less than 1 nm, thus achieving 1 order of magnitude higher sunlight absorption than GaAs and Si. We further study PV devices based on just two stacked monolayers: (1) a Schottky barrier solar cell between MoS2 and graphene and (2) an excitonic solar cell based on a MoS2/WS2 bilayer. We demonstrate that such 1 nm thick active layers can attain power conversion efficiencies of up to ∼1%, corresponding to approximately 1–3 orders of magnitude higher power densities than the best existing ultrathin solar cells. Our work shows that two-dimensional monolayer materials hold yet untapped potential for solar energy absorption and conversion at the nanoscale.
Co-reporter:Marco Bernardi and Jeffrey C. Grossman
The Journal of Physical Chemistry C 2013 Volume 117(Issue 51) pp:26896-26904
Publication Date(Web):November 14, 2013
DOI:10.1021/jp4090348
Materials employed to harvest sunlight are commonly recognized to be at a premium when their optical absorption peaks in the visible, extends to the infrared, is panchromatic, and is matched to the solar spectrum. By contrast, natural photosynthetic absorbers such as chlorophylls and carotenoids display absorption spectra with narrow peaks for yet-unknown evolutionary reasons. Beyond such general observations, a rigorous treatment of sunlight harvesting optimization is still lacking. In this work, we provide a quantitative analysis of optimal solar energy harvesting in materials. We derive optimal absorption spectra as a function of absorber thickness, elucidate the concept of solar-matched absorption and its applicability limits, and define a procedure to rank photovoltaic materials for sunlight harvesting. In addition, we suggest a possible explanation for why absorption in plant photosynthetic pigments occurs in narrow energy windows.
Co-reporter:Priyank V. Kumar, Marco Bernardi, and Jeffrey C. Grossman
ACS Nano 2013 Volume 7(Issue 2) pp:1638
Publication Date(Web):January 31, 2013
DOI:10.1021/nn305507p
Reduced graphene oxide (rGO) is a promising material for a variety of thin-film optoelectronic applications. Two main barriers to its widespread use are the lack of (1) fabrication protocols leading to tailored functionalization of the graphene sheet with oxygen-containing chemical groups, and (2) understanding of the impact of such functional groups on the stability and on the optical and electronic properties of rGO. We carry out classical molecular dynamics and density functional theory calculations on a large set of realistic rGO structures to decompose the effects of different functional groups on the stability, work function, and photoluminescence. Our calculations indicate the metastable nature of carbonyl-rich rGO and its favorable transformation to hydroxyl-rich rGO at room temperature via carbonyl-to-hydroxyl conversion reactions near carbon vacancies and holes. We demonstrate a significant tunability in the work function of rGO up to 2.5 eV by altering the composition of oxygen-containing functional groups for a fixed oxygen concentration, and of the photoluminescence emission by modulating the fraction of epoxy and carbonyl groups. Taken together, our results guide the application of tailored rGO structures in devices for optoelectronics and renewable energy.Keywords: density functional theory; functional groups; molecular dynamics; photoluminescence; reduced graphene oxide; stability; work function
Co-reporter:Priyank V. Kumar, Michael P. Short, Sidney Yip, Bilge Yildiz, and Jeffrey C. Grossman
The Journal of Physical Chemistry C 2013 Volume 117(Issue 11) pp:5678-5683
Publication Date(Web):March 7, 2013
DOI:10.1021/jp309434a
Transition metal-doped ferrites are attractive candidates for a wide range of applications including catalysis and electronic and magnetic devices. Although their bulk characteristics are well-understood, very little is known about their surface properties at the molecular level. Here, we demonstrate high reactivity of NiFe2O4 (111) surfaces, a Ni-doped ferrite, by elucidating the surface structure and water adsorption mechanism using density functional theory with on-site correction for Couloumb interaction (DFT + U). The surface reactivity of NiFe2O4 (111) surfaces (with 0.25 ML Fetet1 and 0.5 ML Feoct2–tet1 terminations) is shown to be significantly higher in comparison with the undoped Fe3O4 (111) surfaces. Dissociation of water is found to be highly favorable with an adsorption energy of −1.11 eV on the 0.25 ML Fetet1 terminated surface and −2.30 eV on the 0.5 ML Feoct2–tet1 terminated surface. In addition, we computed a low activation barrier of 0.18 eV for single water molecule dissociation on the 0.25 ML Fetet1 termination, while the corresponding dissociation reaction on the 0.5 ML Feoct2–tet1 termination proceeded without a barrier. The reactivity of NiFe2O4 surfaces toward water is understood based on strong interactions between the adsorbing OH radical molecular orbitals and the d orbitals of the surface Fe atom. In particular, the new bonding orbitals created due to the interaction of the OH 3σ orbital and the Fe d states are pushed deeper down the energy axis resulting in a greater energy gain and higher water adsorption strength in the case of 0.5 ML Feoct2–tet1 termination. Furthermore, transition-metal surface resonances (TMSR) are found to be good descriptors of the surface reactivity in the two ferrites investigated and is a useful measure to design ferrite-based catalytic systems. These findings have strong implications toward the use of NiFe2O4 as an effective metal-doped ferrite catalyst in a typical industrial process such as the water-gas shift (WGS) reaction and are of significance in fuel materials durability in nuclear reactors where ferrites are known to trap boron resulting in failure of the reactors.
Co-reporter:E. Durgun and Jeffrey C. Grossman
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 6) pp:854-860
Publication Date(Web):March 4, 2013
DOI:10.1021/jz301877n
Solar-thermal fuels reversibly store solar energy in the chemical bonds of molecules by photoconversion, and can release this stored energy in the form of heat upon activation. Many conventional photoswichable molecules could be considered as solar thermal fuels, although they suffer from low energy density or short lifetime in the photoinduced high-energy metastable state, rendering their practical use unfeasible. We present a new approach to the design of chemistries for solar thermal fuel applications, wherein well-known photoswitchable molecules are connected by different linker agents to form molecular rings. This approach allows for a significant increase in both the amount of stored energy per molecule and the stability of the fuels. Our results suggest a range of possibilities for tuning the energy density and thermal stability as a function of the type of the photoswitchable molecule, the ring size, or the type of linkers.Keywords: azobenzene; density functional theory; photoswitch; renewable energy; solar thermal fuel;
Co-reporter:Marco Bernardi, Nicola Ferralis, Jin H. Wan, Rachelle Villalon and Jeffrey C. Grossman
Energy & Environmental Science 2012 vol. 5(Issue 5) pp:6880-6884
Publication Date(Web):08 Mar 2012
DOI:10.1039/C2EE21170J
We formulate, solve computationally and study experimentally the problem of collecting solar energy in three dimensions. We demonstrate that absorbers and reflectors can be combined in the absence of sun tracking to build three-dimensional photovoltaic (3DPV) structures that can generate measured energy densities (energy per base area, kWh/m2) higher by a factor of 2–20 than stationary flat PV panels for the structures considered here, compared to an increase by a factor of 1.3–1.8 for a flat panel with dual-axis sun tracking. The increased energy density is countered by a larger solar cell area per generated energy for 3DPV compared to flat panels (by a factor of 1.5–4 in our conditions), but accompanied by a vast range of improvements. 3DPV structures can mitigate some of the variability inherent to solar PV as they provide a more even source of solar energy generation at all latitudes: they can double the number of peak power generation hours and dramatically reduce the seasonal, latitude and weather variations of solar energy generation compared to a flat panel design. Self-supporting 3D shapes can create new schemes for PV installation and the increased energy density can facilitate the use of cheaper thin film materials in area-limited applications. Our findings suggest that harnessing solar energy in three dimensions can open new avenues towards Terawatt-scale generation.
Co-reporter:David Cohen-Tanugi and Jeffrey C. Grossman
Nano Letters 2012 Volume 12(Issue 7) pp:3602-3608
Publication Date(Web):June 5, 2012
DOI:10.1021/nl3012853
We show that nanometer-scale pores in single-layer freestanding graphene can effectively filter NaCl salt from water. Using classical molecular dynamics, we report the desalination performance of such membranes as a function of pore size, chemical functionalization, and applied pressure. Our results indicate that the membrane’s ability to prevent the salt passage depends critically on pore diameter with adequately sized pores allowing for water flow while blocking ions. Further, an investigation into the role of chemical functional groups bonded to the edges of graphene pores suggests that commonly occurring hydroxyl groups can roughly double the water flux thanks to their hydrophilic character. The increase in water flux comes at the expense of less consistent salt rejection performance, which we attribute to the ability of hydroxyl functional groups to substitute for water molecules in the hydration shell of the ions. Overall, our results indicate that the water permeability of this material is several orders of magnitude higher than conventional reverse osmosis membranes, and that nanoporous graphene may have a valuable role to play for water purification.
Co-reporter:E. Durgun, H. Manzano, R. J. M. Pellenq, and Jeffrey C. Grossman
Chemistry of Materials 2012 Volume 24(Issue 7) pp:1262
Publication Date(Web):March 29, 2012
DOI:10.1021/cm203127m
First principles calculations are employed to provide a fundamental understanding of the relationship between the reactivity of synthetic calcium silicate phases and their electronic structure. Our aim is to shed light on the wide range of hydration kinetics observed in different phases of calcium silicate. For example, while the dicalcium silicate (Ca2SiO4) phase slowly reacts with water, the tricalcium silicate (Ca3SiO5) shows much faster hydration kinetics. We show that the high reactivity of Ca3SiO5 is mainly related to the reactive sites around its more ionic oxygen atoms. Ca2SiO4 does not contain these types of oxygen atoms, although experiments suggest that impurities may play a role in changing the reactivity of these materials. We analyze the electronic structure of a wide range of possible substitutions in both Ca3SiO5 and Ca2SiO4 and show that while the influence of different types of impurities on structural properties is similar, their effect on reactivity is very different. Our calculations suggest that the variation of electronic structure is mainly related to the formation of new hybridized orbitals and the charge exchange between the impurity atoms and the bulk material. The charge localization upon introducing impurities is quantified to predict candidate substitutions that could increase the reactivity of Ca2SiO4, which would broaden the applicability of this lower temperature and thus less costly and energetically less demanding phase.Keywords: calcium silicate; cement clinker; chemical impurity; dissolution; reactivity;
Co-reporter:Priyank V. Kumar ; Michael P. Short ; Sidney Yip ; Bilge Yildiz
The Journal of Physical Chemistry C 2012 Volume 116(Issue 18) pp:10113-10119
Publication Date(Web):May 1, 2012
DOI:10.1021/jp301607h
The present study investigates the adsorption and dissociation reaction pathways of boric acid, B(OH)3, and the reaction kinetic descriptors on NiO(001) and ZrO2(1̅11) surfaces. Density functional theory is employed for ground-state calculations, while the nudged elastic band method is used for obtaining reaction barriers. Strong electron correlations in the case of NiO are included using the DFT + U approach. Adsorption of boric acid on clean ZrO2(1̅11) is found to be more favorable compared with that on NiO(001), in agreement with prior experiments. Dissociative adsorption is observed to dominate over molecular adsorption in the case of ZrO2(1̅11), whereas NiO(001) favors molecular adsorption. The most stable configuration for B(OH)3 on NiO(001) is a hydrogen-bonded molecular structure, Nis-(OH)B(OH)(OH)···Os (s = surface atom), with an adsorption energy of −0.74 eV. On ZrO2(1̅11), a single O–H dissociated structure, Zrs-(O)B(OH)(HO)-Zrs + Os-H, with an adsorption energy of −1.61 eV, is the most stable. Our results reveal lower activation barriers for B(OH)3 dissociation on NiO(001) than on ZrO2(1̅11). We demonstrate the importance of both the surface transition-metal atom and oxygen states and discuss bonding mechanisms leading to different adsorption configurations on such metal oxides. The analysis of surface reactivity presented here is useful in designing metal oxides for catalytic applications and is of significant importance in fuel materials durability in nuclear energy systems.
Co-reporter:Jeong Yun Kim, Joo-Hyoung Lee, and Jeffrey C. Grossman
ACS Nano 2012 Volume 6(Issue 10) pp:9050
Publication Date(Web):September 13, 2012
DOI:10.1021/nn3031595
We investigate the effects of two-dimensional (2D) periodic patterns of functional groups on the thermal transport in a graphene monolayer by employing molecular and lattice dynamics simulations. Our calculations show that the use of patterned 2D shapes on graphene reduces the room temperature thermal conductivity, by as much as 40 times lower than that of the pristine monolayer, due to a combination of boundary and clamping effects. Lattice dynamics calculations elucidate the correlation between this large reduction in thermal conductivity and two dynamical properties of the main heat carrying phonon modes: (1) decreased phonon lifetimes by an order of magnitude due to scattering, and (2) direction-dependent group velocities arising from phonon confinement. Taken together, these results suggest that patterned graphene nanoroads provide a method for tuning the thermal conductivity of graphene without the introduction of defects in the lattice, opening an important possibility for thermoelectric applications.Keywords: chemical functionalization; graphene; lattice dynamics; molecular dynamics; thermal conductivity; thermoelectrics
Co-reporter:Marco Bernardi, Jessica Lohrman, Priyank V. Kumar, Alec Kirkeminde, Nicola Ferralis, Jeffrey C. Grossman, and Shenqiang Ren
ACS Nano 2012 Volume 6(Issue 10) pp:8896
Publication Date(Web):September 6, 2012
DOI:10.1021/nn302893p
Carbon materials are excellent candidates for photovoltaic solar cells: they are Earth-abundant, possess high optical absorption, and maintain superior thermal and photostability. Here we report on solar cells with active layers made solely of carbon nanomaterials that present the same advantages of conjugated polymer-based solar cells, namely, solution processable, potentially flexible, and chemically tunable, but with increased photostability and the possibility to revert photodegradation. The device active layer composition is optimized using ab initio density functional theory calculations to predict type-II band alignment and Schottky barrier formation. The best device fabricated is composed of PC70BM fullerene, semiconducting single-walled carbon nanotubes, and reduced graphene oxide. This active-layer composition achieves a power conversion efficiency of 1.3%—a record for solar cells based on carbon as the active material—and we calculate efficiency limits of up to 13% for the devices fabricated in this work, comparable to those predicted for polymer solar cells employing PCBM as the acceptor. There is great promise for improving carbon-based solar cells considering the novelty of this type of device, the high photostability, and the availability of a large number of carbon materials with yet untapped potential for photovoltaics. Our results indicate a new strategy for efficient carbon-based, solution-processable, thin film, photostable solar cells.Keywords: ab initio materials design; carbon; photodegradation; photovoltaics
Co-reporter:Marco Bernardi, Maurizia Palummo, and Jeffrey C. Grossman
ACS Nano 2012 Volume 6(Issue 11) pp:10082
Publication Date(Web):October 13, 2012
DOI:10.1021/nn303815z
The recent advent of two-dimensional monolayer materials with tunable optical properties and high carrier mobility offers renewed opportunities for efficient, ultrathin excitonic solar cells alternative to those based on conjugated polymer and small molecule donors. Using first-principles density functional theory and many-body calculations, we demonstrate that monolayers of hexagonal BN and graphene (CBN) combined with commonly used acceptors such as PCBM fullerene or semiconducting carbon nanotubes can provide excitonic solar cells with tunable absorber gap, donor–acceptor interface band alignment, and power conversion efficiency, as well as novel device architectures. For the case of CBN–PCBM devices, we predict power conversion efficiency limits in the 10–20% range depending on the CBN monolayer structure. Our results demonstrate the possibility of using monolayer materials in tunable, efficient, ultrathin solar cells in which unexplored exciton and carrier transport regimes are at play.Keywords: band offsets; boron nitride; graphene; monolayer materials; photovoltaics; power conversion efficiency
Co-reporter:Giuseppe Romano;Aldo Di Carlo
Journal of Computational Electronics 2012 Volume 11( Issue 1) pp:8-13
Publication Date(Web):2012 March
DOI:10.1007/s10825-012-0390-2
In this work we compute the effective thermal conductivity of porous Si by means of the phonon Boltzmann transport equation. Simulations of heat transport across aligned square pores reveal that the thermal conductivity can be decreased either by increasing the pore size or decreasing the pore spacing. Furthermore, by including the surface specularity parameter we show that the roughness of the pore walls plays an important role when the pore size is comparable with the phonon mean free path, because to the increase in the surface-to-volume ratio. Thanks to these results, in qualitatively agreement with those obtained with Molecular Dynamics simulations, we gained insights into the scaling of thermal properties of porous materials and interplay between disorder at different length scales. The model, being based on a flexible multiscale finite element context, can be easily integrated with electrical transport models, in order to optimize the figure of merit ZT of thermoelectric devices.
Co-reporter:Alexie M. Kolpak and Jeffrey C. Grossman
Nano Letters 2011 Volume 11(Issue 8) pp:3156-3162
Publication Date(Web):June 20, 2011
DOI:10.1021/nl201357n
Solar thermal fuels, which reversibly store solar energy in molecular bonds, are a tantalizing prospect for clean, renewable, and transportable energy conversion/storage. However, large-scale adoption requires enhanced energy storage capacity and thermal stability. Here we present a novel solar thermal fuel, composed of azobenzene-functionalized carbon nanotubes, with the volumetric energy density of Li-ion batteries. Our work also demonstrates that the inclusion of nanoscale templates is an effective strategy for design of highly cyclable, thermally stable, and energy-dense solar thermal fuels.
Co-reporter:Shenqiang Ren, Marco Bernardi, Richard R. Lunt, Vladimir Bulovic, Jeffrey C. Grossman, and Silvija Gradečak
Nano Letters 2011 Volume 11(Issue 12) pp:5316-5321
Publication Date(Web):October 24, 2011
DOI:10.1021/nl202796u
We demonstrate single-walled carbon nanotube (SWCNT)/P3HT polymer bulk heterojunction solar cells with an AM1.5 efficiency of 0.72%, significantly higher than previously reported (0.05%). A key step in achieving high efficiency is the utilization of semiconducting SWCNTs coated with an ordered P3HT layer to enhance the charge separation and transport in the device active layer. Electrical characteristics of devices with SWCNT concentrations up to 40 wt % were measured and are shown to be strongly dependent on the SWCNT loading. A maximum open circuit voltage was measured for SWCNT concentration of 3 wt % with a value of 1.04 V, higher than expected based on the interface band alignment. Modeling of the open-circuit voltage suggests that despite the large carrier mobility in SWCNTs device power conversion efficiency is governed by carrier recombination. Optical characterization shows that only SWCNT with diameter of 1.3–1.4 nm can contribute to the photocurrent with internal quantum efficiency up to 26%. Our results advance the fundamental understanding and improve the design of efficient polymer/SWCNTs solar cells.
Co-reporter:P. Alex Greaney;Giovanna Lani
Metallurgical and Materials Transactions A 2011 Volume 42( Issue 13) pp:3907-3912
Publication Date(Web):2011 December
DOI:10.1007/s11661-011-0843-4
Surprising Mpemba-like dissipation is observed during computer simulated ring-down of the flexural modes of a single-walled carbon nanotube resonator. Vibrations are made to decay to zero faster by adding a larger initial excitation. We liken this counterintuitive observation to the well-known Mpemba effect in which hot water freezes faster that cold water. In both cases, the system seems to pose a memory of its thermal history; a paradoxical result that is reconciled if the dissipative state of the system is not described uniquely by the system’s average temperature. A vibrational mode projection algorithm is used to track the dissipation pathway, showing that dissipation is dependent strongly on the development of an athermal phonon population. The implications of Mpemba-like behavior in more general, and continuously driven, nanomechanical systems are discussed.
Co-reporter:Yosuke Kanai, Zhigang Wu and Jeffrey C. Grossman
Journal of Materials Chemistry A 2010 vol. 20(Issue 6) pp:1053-1061
Publication Date(Web):01 Oct 2009
DOI:10.1039/B913277P
In this feature article we focus on the key problem of charge separation in nano-scale photovoltaic materials; in particular recent theoretical/computational work based on first principles electronic structure approaches is presented and discussed. We review applications of state-of-the-art electronic structure calculations to nano-scale materials that enable charge separation between an excited electron and hole in so-called excitonic photovoltaic cells. Emphasis is placed on theoretical results that provide insight into experimentally observed processes, which are yet to be understood and do not appear to obey a single unique model but rather depend on atomistic details. Examples are provided that illustrate how computational approaches can be employed to probe new directions in materials design for inducing efficient charge separation. We also discuss the computational challenges in electronic structure theory for reliably predicting and designing new materials suitable for charge separation in photovoltaic applications.
Co-reporter:Marco Bernardi, Michele Giulianini, and Jeffrey C. Grossman
ACS Nano 2010 Volume 4(Issue 11) pp:6599
Publication Date(Web):October 28, 2010
DOI:10.1021/nn1018297
Charge transfer at the interface of conjugated polymer and nanoscale inorganic acceptors is pivotal in determining the efficiency of excitonic solar cells. Despite intense efforts, carbon nanotube/polymer solar cells have resulted in disappointing efficiencies (<2%) due in large part to poor charge transfer at the interface. While the interfacial energy level alignment is clearly important, the self-assembly and the interface structure also play a major role in facilitating this charge transfer. To understand and control this effect to our advantage, we study the interface of commonly used conductive polymer poly-3-hexylthiophene (P3HT) and single-walled carbon nanotubes (SWNTs) with a combination of molecular dynamics simulations, absorption spectra experiments, and an analysis of charge transfer effects. Classical molecular dynamics simulations show that the P3HT wraps around the SWNTs in a number of different conformations, including helices, bundles, and more elongated conformations that maximize planar π−π stacking, in agreement with recent experimental observations. Snapshots from the MD simulations reveal that the carbon nanotubes play an important templating role of increasing the π-conjugation in the system, an effect deriving from the π−π stacking interaction at the interface and the 1-dimensional (1D) nature of the SWNTs, and independent of the SWNT chirality. We show how this increase in the system conjugation could largely improve the charge transfer in P3HT−SWNT type II heterojunctions and support our results with absorption spectra measurements of mixtures of carbon nanotubes and P3HT. These findings open possibilities for improved preparation of polymeric solar cells based on carbon nanotubes and on 1D nanomaterials in general.Keywords: bulk heterojunction; carbon nanotube; charge transfer; conjugation length; excitonic solar cells; molecular dynamics; organic photovoltaics; P3HT; self-assembly
Co-reporter:Dr. Yosuke Kanai;Dr. Varadharajan Srinivasan;Dr. Steven K. Meier;Dr. K. Peter C. Vollhardt; Jeffrey C. Grossman
Angewandte Chemie International Edition 2010 Volume 49( Issue 47) pp:8926-8929
Publication Date(Web):
DOI:10.1002/anie.201002994
Co-reporter:P. Alex Greaney, Giovanna Lani, Giancarlo Cicero and Jeffrey C. Grossman
Nano Letters 2009 Volume 9(Issue 11) pp:3699-3703
Publication Date(Web):October 28, 2009
DOI:10.1021/nl901706y
We observe a new anomalous and transient process of intrinsic dissipation in simulations of the ring-down of flexural modes in single-walled carbon nanotube (CNT) resonators. The effect is pronounced, causing the quality factor of the mode to be reduced by more that 95% for tens of picoseconds. The anomalous dissipation depends on the CNT temperature and the energy in the mode, and remarkably increasing the excitation energy in the resonator causes it to decay to zero faster. By tracking the cascade of energy as it dissipates we identify “gateway” modes that provide important channels for dissipation. The processes we observe show that an athermal phonon population accompanying dissipation can strongly influence the quality factor in nanoelectromechanical devices.
Co-reporter:Zhigang Wu ; Mark D. Allendorf
Journal of the American Chemical Society 2009 Volume 131(Issue 39) pp:13918-13919
Publication Date(Web):September 9, 2009
DOI:10.1021/ja905639m
We calculated the desorption energy of MgH2 clusters using the highly accurate quantum Monte Carlo (QMC) approach, which can provide desorption energies with chemical accuracy (within ∼1 kcal/mol) and therefore provides a valuable benchmark for such hydrogen-storage simulations. Compared with these QMC results, the most widely used density functional theory (DFT) computations (including a wide range of exchange-correlation functionals) cannot reach a consistent and suitable level of accuracy across the thermodynamically tunable range for MgH2 clusters. Furthermore, our QMC calculations show that the DFT error depends substantially on cluster size. These results suggest that in simulating metal−hydride systems it is very important to apply accurate methods that go beyond traditional mean-field approaches as a benchmark of their performance for a given material, and QMC is an appealing method to provide such a benchmark due to its high level of accuracy and favorable scaling (N3) with the number of electrons.
Co-reporter:David Cohen-Tanugi, Jeffrey C. Grossman
Desalination (15 June 2015) Volume 366() pp:59-70
Publication Date(Web):15 June 2015
DOI:10.1016/j.desal.2014.12.046
•We review recent progress in the computational study of graphene as an RO membrane.•We introduce graphene and current knowledge about its mass transport properties.•We examine six key mechanisms that govern salt rejection in graphene.•Molecular dynamics have played a dominant role in the study of graphene membranes.•We suggest a greater role for quantum-level simulations and macroscale computation.In this review, we examine the potential and the challenges of designing an ultrathin reverse osmosis (RO) membrane from graphene, focusing on the role of computational methods in designing, understanding, and optimizing the relationship between atomic structure and RO performance. In recent years, graphene has emerged as a promising material for improving the performance of RO. Beginning at the atomic scale and extending to the RO plant scale, we review applications of computational research that have explored the structure, properties and potential performance of nanoporous graphene in the context of RO desalination.Download full-size image
Co-reporter:Li-Chiang Lin, Jongwon Choi and Jeffrey C. Grossman
Chemical Communications 2015 - vol. 51(Issue 80) pp:NaN14924-14924
Publication Date(Web):2015/08/14
DOI:10.1039/C5CC05969K
We computationally demonstrate that two-dimensional covalent triazine frameworks (CTFs) provide opportunities in water desalination. By varying the chemical building blocks, the pore structure, chemistry, and membrane performance can be designed, leading to two orders of magnitude higher water permeability than polyamide membranes while maintaining excellent ability to reject salts.
Co-reporter:Yosuke Kanai, Zhigang Wu and Jeffrey C. Grossman
Journal of Materials Chemistry A 2010 - vol. 20(Issue 6) pp:NaN1061-1061
Publication Date(Web):2009/10/01
DOI:10.1039/B913277P
In this feature article we focus on the key problem of charge separation in nano-scale photovoltaic materials; in particular recent theoretical/computational work based on first principles electronic structure approaches is presented and discussed. We review applications of state-of-the-art electronic structure calculations to nano-scale materials that enable charge separation between an excited electron and hole in so-called excitonic photovoltaic cells. Emphasis is placed on theoretical results that provide insight into experimentally observed processes, which are yet to be understood and do not appear to obey a single unique model but rather depend on atomistic details. Examples are provided that illustrate how computational approaches can be employed to probe new directions in materials design for inducing efficient charge separation. We also discuss the computational challenges in electronic structure theory for reliably predicting and designing new materials suitable for charge separation in photovoltaic applications.
Co-reporter:Ggoch Ddeul Han, Sarah S. Park, Yun Liu, David Zhitomirsky, Eugene Cho, Mircea Dincă and Jeffrey C. Grossman
Journal of Materials Chemistry A 2016 - vol. 4(Issue 41) pp:NaN16165-16165
Publication Date(Web):2016/09/13
DOI:10.1039/C6TA07086H
Photocontrolled self-assembly of molecules has been utilized to change the physical properties of organic materials for various applications, while photon energy storage materials that incorporate photochromic molecules such as azobenzenes have been recognized as another highly attractive class of materials that convert and store photon energy in the strained chemical bonds. Herein, we demonstrate the photocontrolled self-assembly and disassembly of photon energy storage materials based on new diacetylene derivatives with azobenzene moieties and with varied alkyl spacers and linkers. We developed a series of symmetric diacetylenes and polydiacetylenes and obtained high energy-density materials that can store up to 176.2 kJ mol−1 (or 200.2 kJ mol−1, if completely charged); more than double that of pristine azobenzene. The extra energy storage in the materials in addition to the isomerization enthalpy of azobenzene units is enabled by the different phase of materials in the ground state (crystalline solid) and in metastable state (amorphous solid/liquid). It is notable that the phase change characteristic of organic materials can be a parameter to consider in terms of designing high energy density photon energy storage materials.
.jpg)