Co-reporter:Uttio Roy Chowdhury, Peter I. Dosa, Michael P. Fautsch
Experimental Eye Research 2017 Volume 158(Volume 158) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.exer.2016.04.020
•ATP sensitive potassium channels contain sulfonylurea receptor (SUR) and potassium inward rectifying (Kir) subunits.•ATP sensitive potassium channels are found in many tissues of the body including the trabecular meshwork and retina.•ATP sensitive potassium channel openers show ocular hypotensive activity in human anterior segment cultures and in mice.•ATP sensitive potassium channels have direct neuroprotective properties on retinal ganglion cells.ATP sensitive potassium (KATP) channels connect the metabolic and energetic state of cells due to their sensitivity to ATP and ADP concentrations. KATP channels have been identified in multiple tissues and organs of the body including heart, pancreas, vascular smooth muscles and skeletal muscles. These channels are obligatory hetero-octamers and contain four sulfonylurea (SUR) and four potassium inward rectifier (Kir) subunits. Based on the particular type of SUR and Kir present, there are several tissue specific subtypes of KATP channels, each with their own unique set of functions. Recently, KATP channels have been reported in human and mouse ocular tissues. In ex vivo and in vivo model systems, KATP channel openers showed significant ocular hypotensive properties with no appearance of toxic side effects. Additionally, when used in conjunction with known intraocular pressure lowering drugs, an additive effect on IOP reduction was observed. These KATP channel openers have also been reported to protect the retinal ganglion cells during ischemic stress and glutamate induced toxicity suggesting a neuroprotective property for this drug class. Medications that are currently used for treating ocular hypertensive diseases like glaucoma do not directly protect the affected retinal cells, are sometimes ineffective and may show significant side effects. In light of this, KATP channel openers with both ocular hypotensive and neuroprotective properties, have the potential to develop into a new class of glaucoma therapeutics.
Co-reporter:Uttio Roy Chowdhury; Kimberly B. Viker; Kristen L. Stoltz; Bradley H. Holman; Michael P. Fautsch;Peter I. Dosa
Journal of Medicinal Chemistry 2016 Volume 59(Issue 13) pp:6221-6231
Publication Date(Web):July 1, 2016
DOI:10.1021/acs.jmedchem.6b00406
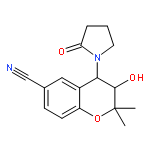
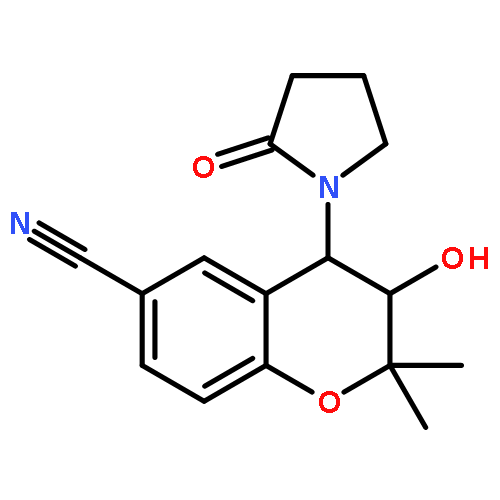
ATP-sensitive potassium (KATP) channel openers have emerged as potential therapeutics for the treatment of glaucoma, lowering intraocular pressure (IOP) in animal models and cultured human anterior segments. We have prepared water-soluble phosphate and dipeptide derivatives of the KATP channel opener cromakalim and evaluated their IOP lowering capabilities in vivo. In general, the phosphate derivatives proved to be more chemically robust and efficacious at lowering IOP with once daily dosing in a normotensive mouse model. Two of these phosphate derivatives were further evaluated in a normotensive rabbit model, with a significant difference in activity observed. No toxic effects on cell structure or alterations in morphology of the aqueous humor outflow pathway were observed after treatment with the most efficacious compound, (3S,4R)-2, suggesting that it is a strong candidate for development as an ocular hypotensive agent.
Co-reporter:Peter I. Dosa;Elizabeth Ambrose Amin
Journal of Medicinal Chemistry 2016 Volume 59(Issue 3) pp:810-840
Publication Date(Web):September 21, 2015
DOI:10.1021/acs.jmedchem.5b00982
There are many reported examples of small structural modifications to GPCR-targeted ligands leading to major changes in their functional activity, converting agonists into antagonists or vice versa. These shifts in functional activity are often accompanied by negligible changes in binding affinity. The current perspective focuses on outlining and analyzing various approaches that have been used to interconvert GPCR agonists, partial agonists, and antagonists in order to achieve the intended functional activity at a GPCR of therapeutic interest. An improved understanding of specific structural modifications that are likely to alter the functional activity of a GPCR ligand may be of use to researchers designing GPCR-targeted drugs and/or probe compounds, specifically in cases where a particular ligand exhibits good potency but not the preferred functional activity at the GPCR of choice.
Co-reporter:Peter I. Dosa, Tim Ward, Michael A. Walters, and Suck Won Kim
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 2) pp:254
Publication Date(Web):January 14, 2013
DOI:10.1021/ml3003814
The dopamine agonist cabergoline has been used to treat prolactinomas, Parkinson’s disease, Cushing’s disease, and sexual dysfunction. However, its clinical use was severely curtailed when it was found that patients taking cabergoline had an increased risk of developing cardiac-valve regurgitation. This potentially life-threatening condition has been associated with drugs, such as cabergoline, that are 5-HT2B receptor agonists. We prepared analogs of cabergoline and have identified several that have limited or no agonism at the 5-HT2B receptor.Keywords: 5-HT2B; Cabergoline; dopamine agonist; ergot alkaloid; sexual dysfunction
Co-reporter:Dr. Peter I. Dosa;Dr. Tim Ward;Dr. Rui E. Castro;Dr. Cecília M. P. Rodrigues;Dr. Clifford J. Steer
ChemMedChem 2013 Volume 8( Issue 6) pp:1002-1011
Publication Date(Web):
DOI:10.1002/cmdc.201300059
Abstract
Ursodeoxycholic acid (UDCA) is a bile acid with demonstrated anti-apoptotic activity in both in vitro and in vivo models. However, its utility is hampered by limited aqueous solubility. As such, water-soluble prodrugs of UDCA could have an advantage over the parent bile acid in indications where intravenous administration might be preferable, such as decreasing damage from stroke or acute kidney injury. Five phosphate prodrugs were synthesized, including one incorporating a novel phosphoryloxymethyl carboxylate (POMC) moiety. These prodrugs were highly water-soluble, but showed significant differences in chemical stability, with oxymethylphosphate prodrugs being the most unstable. In a series of NMR experiments, the POMC prodrug was bioactivated to UDCA by alkaline phosphatase (AP) faster than a prodrug containing a phosphate directly attached to the alcohol at the 3-position of UDCA. Both of these prodrugs showed significant anti-apoptotic activity in a series of in vitro assays, although the POMC prodrug required the addition of AP for activity, while the other compound was active without exogenous AP.
Co-reporter:Peter I. Dosa, Sonja Strah-Pleynet, Honnappa Jayakumar, Martin Casper, Marc Decaire, Yifeng Xiong, Juerg Lehmann, Karoline Choi, Katie Elwell, Amy Wong, Robert R. Webb, John W. Adams, Juan Ramirez, Jeremy G. Richman, William Thomsen, Graeme Semple, Bradley R. Teegarden
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 18) pp:5486-5489
Publication Date(Web):15 September 2009
DOI:10.1016/j.bmcl.2009.07.073
Potent 5-HT2A inverse-agonists containing phenyl-pyrazole ureas with an amino side chain were identified. Optimization of this series resulted in selective compounds that proved effective in modulating 5HT-induced amplification of ADP-stimulated human platelet aggregation.

![2-[[(4r)-4-[(3r,5s,7r,8r,9s,10s,12s,13r,14s,17r)-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]ethanesulfonic Acid](http://img.cochemist.com/ccimg/100/81-24-3.png)
![2-[[(4r)-4-[(3r,5s,7r,8r,9s,10s,12s,13r,14s,17r)-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]ethanesulfonic Acid](http://img.cochemist.com/ccimg/100/81-24-3_b.png)