Co-reporter:Yi Yu, Jianfen Fan, Xiliang Yan, Jian Xu, and Mingming Zhang
The Journal of Physical Chemistry A 2015 Volume 119(Issue 20) pp:4723-4734
Publication Date(Web):April 24, 2015
DOI:10.1021/acs.jpca.5b01380
MD simulations have been carried out for studying the tilt behaviors of the 8 × (WL)4-CPNT embedded in the POPE bilayer in a water environment and under an anhydrous condition, respectively. Besides, the dependences of the transport characteristics of channel water on the extent of the CPNT tilt were explored. The results indicate that the presence of water may exacerbate the CPNT tilt but plays an important role in maintaining the integrity of the CPNT by forming water bridges in the end-gaps. Cation−π interactions between the indole rings of Trp residues and lipid headgroups are the major factor causing the CPNT tilt under an anhydrous condition, while H-bonded interactions between water molecules and the indole rings are primary in a water environment. The dipole orientations of channel water molecules except those in the last end-gap are gradually oriented and eventually only take “+dipole” states under 20° of the CPNT tilt. The average residence time of channel water gradually increases with the intensifying of the CPNT tilt.
Co-reporter:Rui Li, Jianfen Fan, Hui Li, Xiliang Yan, and Yi Yu
The Journal of Physical Chemistry B 2013 Volume 117(Issue 48) pp:14916-14927
Publication Date(Web):November 18, 2013
DOI:10.1021/jp408769u
The dynamic behaviors and transport properties of O2, CO2, and NH3 molecules through a transmembrane cyclic peptide nanotube (CPNT) of 8×cyclo-(WL)4/POPE have been investigated by steered molecular dynamics (SMD) simulations and adaptive biasing force (ABF) samplings. Different external forces are needed for three gas molecules to enter the channel. The periodic change of the pulling force curve for a gas traveling through the channel mainly arises from the regular and periodic arrangement of the composed CP subunits of the CPNT. Radial distribution functions (RDFs) between gas and water disclose the density decrease of channel water, which strongly aggravates the discontinuity of H-bond formation between a gas molecule and the neighboring water. Compared to hardly any H-bond formation between CO2 (or O2) and the framework of the CPNT, NH3 can form abundant H-bonds with the carbonyl/amide groups of the CPNT, leading to a fierce competition to NH3–water H-bonded interactions. In addition to direct H-bonded interactions, all three gases can form water bridges with the tube. The potential profile of mean force coincides with the occurring probability of a gas molecule along the tube axis. The energy barriers at two mouths of the CPNT elucidate the phenomenon that CO2 and O2 are thoroughly confined in the narrow lumen while NH3 can easily go outside the tube. Intermolecular interactions of each gas with channel water and the CPNT framework and the formation of H-bonds and water bridges illuminate the different gas translocation behaviors. The results uncover interesting and comprehensive mechanisms underlying the permeation characteristics of three gas molecules traveling through a transmembrane CPNT.
Co-reporter:Min Cen;Jian Fen Fan;Dong Yan Liu;Xue Zeng Song
Journal of Molecular Modeling 2013 Volume 19( Issue 2) pp:601-611
Publication Date(Web):2013 February
DOI:10.1007/s00894-012-1588-8
Molecular dynamic (MD) simulations have been performed to study the behaviors of ten kinds of cyclo-hexa-peptides (CHPs) composed of amino acids with the diverse hydrophilic/hydrophobic side chains at the water/cyclohexane interface. All the CHPs take the “horse-saddle” conformations at the interface and the hydrophilicity/hydrophobicity of the side chains influences the backbones’ structural deformations. The orientations and distributions of the CHPs at the interface and the differences of interaction energies (ΔΔE) between the CHPs and the two liquid phases have been determined. RDF analysis shows that the H-bonds were formed between the OC atoms of the CHPs’ backbones and Hw atoms of water molecules. N atoms of the CHPs’ backbones formed the H-bonds or van der Waals interactions with the water solvent. It was found that there is a parallel relationship between ΔΔE and the lateral diffusion coefficients (Dxy) of the CHPs at the interface. The movements of water molecules close to the interface are confined to some extent, indicating that the dynamics of the CHPs and interfacial water molecules are strongly coupled.
Co-reporter:Jian Liu, Jianfen Fan, Min Cen, Xuezeng Song, Dongyan Liu, Weiqun Zhou, Zhao Liu, and Jianfeng Yan
Journal of Chemical Information and Modeling 2012 Volume 52(Issue 8) pp:2132-2138
Publication Date(Web):July 26, 2012
DOI:10.1021/ci300185c
Effects of the channel length and membrane thickness on the water permeation through the transmembrane cyclic octa-peptide nanotubes (octa-PNTs) have been studied by molecular dynamics (MD) simulations. The water osmotic permeability (pf) through the PNTs of k × (WL)4/POPE (1-palmitoyl-2-oleoyl-glycerophosphoethanolamine; k = 6, 7, 8, 9, and 10) was found to decay with the channel length (L) along the axis (∼L–2.0). Energetic analysis showed that a series of water binding sites exist in these transmembrane PNTs, with the barriers of ∼3kBT, which elucidates the tendency of pf well. Water diffusion permeability (pd) exhibits a relationship of ∼L–1.8, which results from the novel 1–2–1–2 structure of water chain in such confined nanolumens. In the range of simulation accuracy, the ratio (pf/pd) of the water osmotic and diffusion permeability is approximately a constant. MD simulations of water permeation through the transmembrane PNTs of 8 × (WL)4/octane with the different octane membrane thickness revealed that the water osmotic and diffusion permeability (pf and pd) are both independent of the octane membrane thickness, confirmed by the weak and nearly same interactions between the channel water and octane membranes with the different thickness. The results may be helpful for revealing the permeation mechanisms of biological water channels and designing artificial nanochannels.
Co-reporter:Jian-Fen Fan;Li-Fen Wu;Yun-Peng Sun
Chinese Journal of Chemistry 2007 Volume 25(Issue 4) pp:
Publication Date(Web):5 APR 2007
DOI:10.1002/cjoc.200790089
DFT-B3LYP calculations were carried out to study the enantioselectivity of the (S)-4-hydroxylproline-catalyzed direct aldol reaction between acetone and 4-nitrobenzaldehyde. Four transition structures associated with the stereo-controlling step of the reaction have been determined. They are corresponding to the anti and syn arrangements of the methylene moiety related to the carboxylic acid group in enamine intermediate and the si and re attacks to the aldehyde carbonyl carbon. The effect of DMSO solvent on the stereo-controlling step was investigated with polarized continuum model (PCM). The computed energies of the transition states reveal the moderate enantioselectivity of the reaction.

![Benzamide, 2-fluoro-N-[[(2-methylphenyl)amino]thioxomethyl]-](/data/chemimg/135700/536722-20-0.png)
![Benzamide, 2-fluoro-N-[[(2-methylphenyl)amino]thioxomethyl]-](/data/chemimg/135700/536722-20-0_b.png)
![Benzamide, 4-fluoro-N-[[(4-methylphenyl)amino]thioxomethyl]-](/data/chemimg/135700/446831-62-5.png)
![Benzamide, 4-fluoro-N-[[(4-methylphenyl)amino]thioxomethyl]-](/data/chemimg/135700/446831-62-5_b.png)
![Benzamide, N-[[[4-(dimethylamino)phenyl]amino]thioxomethyl]-2-fluoro-](/data/chemimg/135700/425682-41-3.png)
![Benzamide, N-[[[4-(dimethylamino)phenyl]amino]thioxomethyl]-2-fluoro-](/data/chemimg/135700/425682-41-3_b.png)
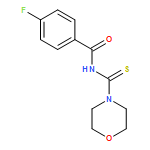
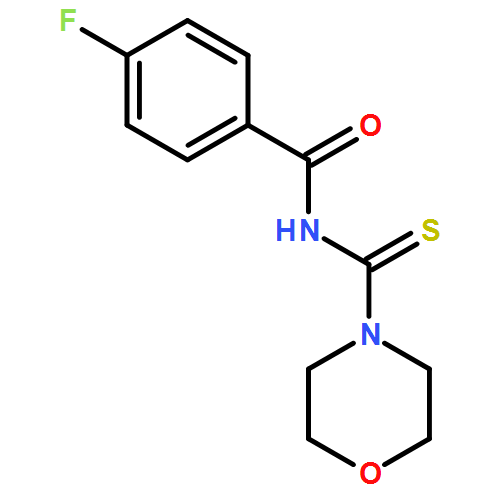
![Benzamide, N-[[[4-(dimethylamino)phenyl]amino]thioxomethyl]-4-fluoro-](/data/chemimg/135700/335212-90-3.png)
![Benzamide, N-[[[4-(dimethylamino)phenyl]amino]thioxomethyl]-4-fluoro-](/data/chemimg/135700/335212-90-3_b.png)
![Benzamide, 2-fluoro-N-[[(4-methylphenyl)amino]thioxomethyl]-](/data/chemimg/135700/156005-06-0.png)
![Benzamide, 2-fluoro-N-[[(4-methylphenyl)amino]thioxomethyl]-](/data/chemimg/135700/156005-06-0_b.png)




![9-Octadecenoic acid(9Z)-,1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexadecyl)oxy]ethylester](http://img.cochemist.com/ccimg/10100/10015-88-0.png)
![9-Octadecenoic acid(9Z)-,1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexadecyl)oxy]ethylester](http://img.cochemist.com/ccimg/10100/10015-88-0_b.png)
![9-Octadecenoic acid(9Z)-,(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexadecyl)oxy]ethylester](http://img.cochemist.com/ccimg/26700/26662-94-2.png)
![9-Octadecenoic acid(9Z)-,(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-2-[(1-oxohexadecyl)oxy]ethylester](http://img.cochemist.com/ccimg/26700/26662-94-2_b.png)